
Quest for Quality in Translational Stroke Research—A New Dawn for Neuroprotection?


Abstract
:1. Introduction
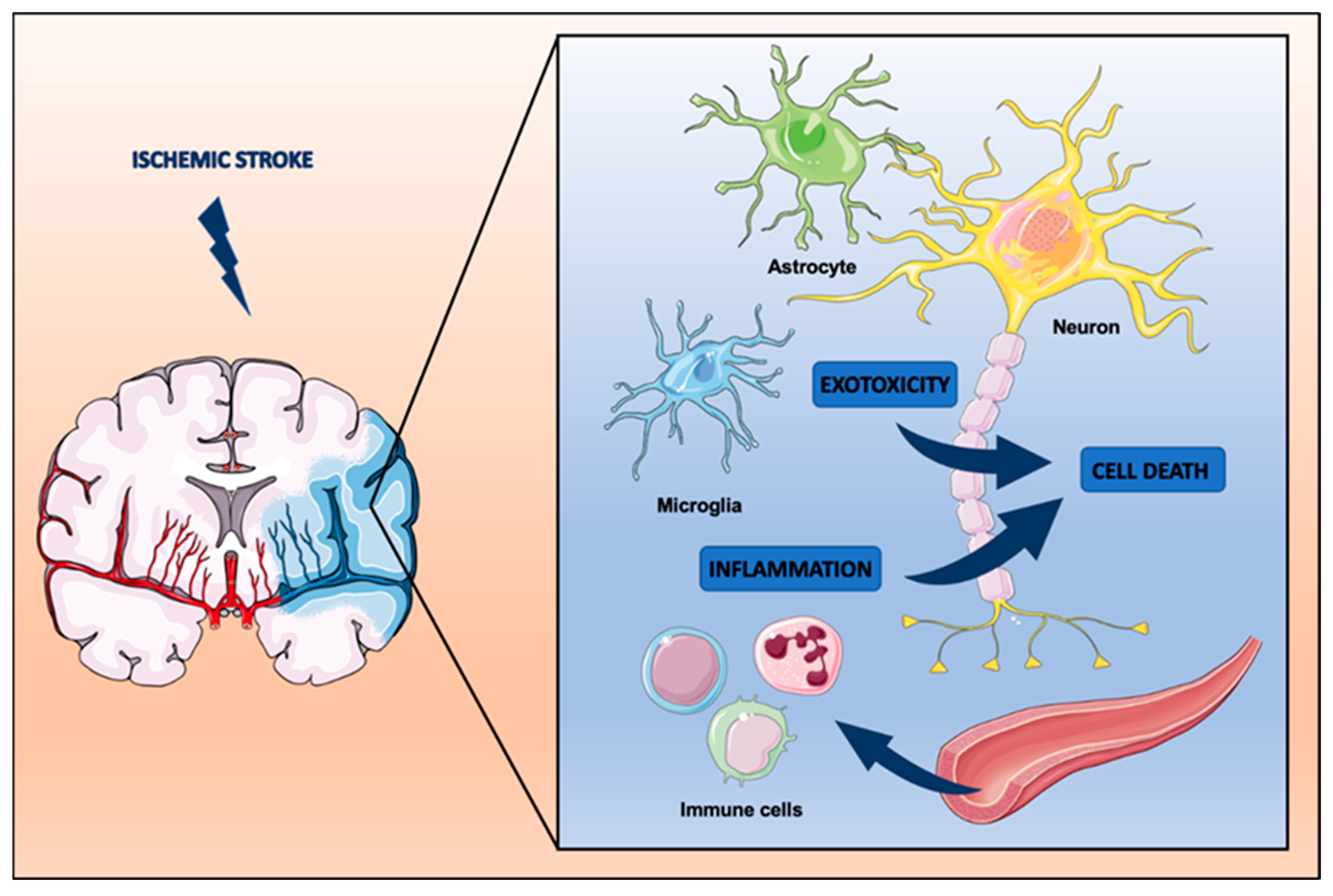
2. Pathophysiology of Stroke
3. Preclinical Studies—A Brief Overview
4. Translational Stroke Studies—A History of Failure
5. Challenges and Perspectives for Bench-to-Bedside Translation
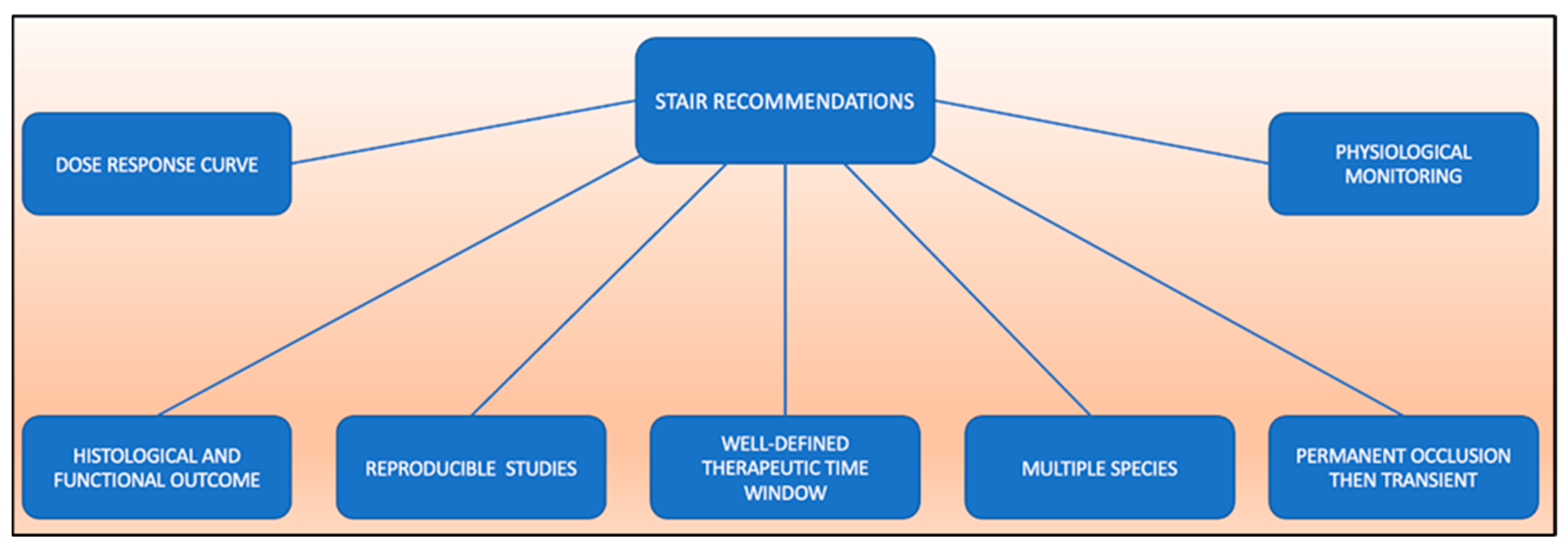
5.1. STAIR Recommendations
5.2. Animal Models
5.3. Occlusion Techniques
5.4. Therapeutic Time Window
6. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CBF | cerebral blood flow |
| ICA | internal carotid artery |
| CCA | common carotid artery |
| MCA | middle cerebral artery |
References
- Donkor, E.S. Stroke in the 21st Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abedi, V.; et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Ginsberg, M.D. Neuroprotection for ischemic stroke: Past, present and future. Neuropharmacology 2008, 55, 363–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, M.; Feuerstein, G.; Howells, D.; Hurn, P.D.; Kent, T.; Savitz, S.I.; Lo, E.H. Update of the Stroke Therapy Academic Industry Roundtable Preclinical Recommendations. Stroke 2009, 40, 2244–2250. [Google Scholar] [CrossRef]
- Roundtable, S.T.A.I. Recommendations for Standards Regarding Preclinical Neuroprotective and Restorative Drug Development. Stroke 1999, 30, 2752–2758. [Google Scholar] [CrossRef]
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.-Q.; Ma, X.-T.; Hu, Z.-W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.-J.; Tian, D.-S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef]
- Planas, A.M. Role of Immune Cells Migrating to the Ischemic Brain. Stroke 2018, 49, 2261–2267. [Google Scholar] [CrossRef]
- Steinberg, G.K.; Kondziolka, D.; Wechsler, L.R.; Lunsford, L.D.; Kim, A.S.; Johnson, J.N.; Bates, D.; Poggio, G.; Case, C.; McGrogan, M.; et al. Two-year safety and clinical outcomes in chronic ischemic stroke patients after implantation of modified bone marrow–derived mesenchymal stem cells (SB623): A phase 1/2a study. J. Neurosurg. 2018, 131, 1462–1472. [Google Scholar] [CrossRef]
- Narayan, S.K.; Cherian, S.G.; Phaniti, P.B.; Chidambaram, S.B.; Vasanthi, A.H.R.; Arumugam, M. Preclinical animal studies in ischemic stroke: Challenges and some solutions. Anim. Model. Exp. Med. 2021, 4, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Bang, O.Y.; Kim, E.H. Mesenchymal Stem Cell-Derived Extracellular Vesicle Therapy for Stroke: Challenges and Progress. Front. Neurol. 2019, 10, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonic, A.; Dottori, M.; Macleod, M.R.; Donnan, G.A.; Howells, D.W. NXY-059, a Failed Stroke Neuroprotectant, Offers No Protection to Stem Cell-Derived Human Neurons. J. Stroke Cerebrovasc. Dis. 2018, 27, 2158–2165. [Google Scholar] [CrossRef]
- Secades, J.J.; Alvarez-Sabin, J.; Castillo, J.; Díez-Tejedor, E.; Martinez-Vila, E.; Ríos, J.; Oudovenko, N. Citicoline for Acute Ischemic Stroke: A Systematic Review and Formal Meta-analysis of Randomized, Double-Blind, and Placebo-Controlled Trials. J. Stroke Cerebrovasc. Dis. 2016, 25, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, A.; Giralt, D.; Garcia-Bonilla, L.; Campos, M.; Rosell, A.; Montaner, J. Citicoline in pre-clinical animal models of stroke: A meta-analysis shows the optimal neuroprotective profile and the missing steps for jumping into a stroke clinical trial. J. Neurochem. 2012, 123, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.-T.; Salter, M.W.; Tymianski, M. Treatment of Ischemic Brain Damage by Perturbing NMDA Receptor- PSD-95 Protein Interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Sun, H.-S.; Doucette, T.A.; Liu, Y.; Fang, Y.; Teves, L.; Aarts, M.; Ryan, C.L.; Bernard, P.B.; Lau, A.; Forder, J.P.; et al. Effectiveness of PSD95 Inhibitors in Permanent and Transient Focal Ischemia in the Rat. Stroke 2008, 39, 2544–2553. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.J.; Teves, L.; Tymianski, M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nat. 2012, 483, 213–217. [Google Scholar] [CrossRef]
- Imai, T.; Iwata, S.; Miyo, D.; Nakamura, S.; Shimazawa, M.; Hara, H. A novel free radical scavenger, NSP-116, ameliorated the brain injury in both ischemic and hemorrhagic stroke models. J. Pharmacol. Sci. 2019, 141, 119–126. [Google Scholar] [CrossRef]
- Yi, N.-X.; Zhou, L.-Y.; Wang, X.-Y.; Song, Y.-J.; Han, H.-H.; Zhang, T.-S.; Wang, Y.-J.; Shi, Q.; Xu, H.; Liang, Q.-Q.; et al. MK-801 attenuates lesion expansion following acute brain injury in rats: A meta-analysis. Neural Regen. Res. 2019, 14, 1919–1931. [Google Scholar] [CrossRef]
- Albers, G.W.; Goldstein, L.B.; Hall, D.; Lesko, L.M. For the Aptiganel Acute Stroke Investigators Aptiganel Hydrochloride in Acute Ischemic Stroke: A randomized controlled trial. JAMA 2001, 286, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Hoyte, L.; Barber, P.A.; Buchan, A.; Hill, M. The Rise and Fall of NMDA Antagonists for Ischemic Stroke. Curr. Mol. Med. 2004, 4, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Hudobenko, J.; Verma, R.; McCullough, L. Abstract TP270: Interleukin-6 Receptor Inhibition with Tocilizumab Ameliorates Ischemic Stroke Damage in Mice. Stroke 2017, 48. [Google Scholar] [CrossRef]
- Fukuta, T.; Asai, T.; Yanagida, Y.; Namba, M.; Koide, H.; Shimizu, K.; Oku, N. Combination therapy with liposomal neuroprotectants and tissue plasminogen activator for treatment of ischemic stroke. FASEB J. 2017, 31, 1879–1890. [Google Scholar] [CrossRef] [Green Version]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Bürkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef]
- Dabrowska, S.; Andrzejewska, A.; Lukomska, B.; Janowski, M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J. Neuroinflamm. 2019, 16, 178. [Google Scholar] [CrossRef] [Green Version]
- Doeppner, T.R.; Bähr, M.; Hermann, D.M.; Giebel, B. Concise Review: Extracellular Vesicles Overcoming Limitations of Cell Therapies in Ischemic Stroke. Stem Cells Transl. Med. 2017, 6, 2044–2052. [Google Scholar] [CrossRef]
- Zheng, M.X.; Haupt, B.M.; Bähr, M.; Tatenhorst, L.; Doeppner, T.R. Treating Cerebral Ischemia: Novel Therapeutic Strategies from Experimental Stroke Research. Cereb. Ischemia 2021, 165–186. [Google Scholar] [CrossRef]
- Haupt, M.; Zheng, X.; Kuang, Y.; Lieschke, S.; Janssen, L.; Bosche, B.; Jin, F.; Hein, K.; Kilic, E.; Venkataramani, V.; et al. Lithium modulates miR-1906 levels of mesenchymal stem cell-derived extracellular vesicles contributing to poststroke neuroprotection by toll-like receptor 4 regulation. Stem Cells Transl. Med. 2020, 10, 357–373. [Google Scholar] [CrossRef]
- Xin, H.; Liu, Z.; Buller, B.; Li, Y.; Golembieski, W.; Gan, X.; Wang, F.; Lu, M.; Ali, M.M.; Zhang, Z.G.; et al. MiR-17-92 enriched exosomes derived from multipotent mesenchymal stromal cells enhance axon-myelin remodeling and motor electrophysiological recovery after stroke. J. Cereb. Blood Flow Metab. 2020, 41, 1131–1144. [Google Scholar] [CrossRef]
- Matsumoto, J.; Stewart, T.; Banks, W.A.; Zhang, J. The Transport Mechanism of Extracellular Vesicles at the Blood-Brain Barrier. Curr. Pharm. Des. 2018, 23, 6206–6214. [Google Scholar] [CrossRef] [PubMed]
- Sena, E.; Wheble, P.; Sandercock, P.; Macleod, M. Systematic Review and Meta-Analysis of the Efficacy of Tirilazad in Experimental Stroke. Stroke 2007, 38, 388–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, H.E.C. A Randomized Trial of Tirilazad Mesylate in Patients With Acute Stroke (RANTTAS). The RANTTAS Investigators. Stroke 1996, 27, 1453–1458. [Google Scholar] [CrossRef]
- Haley, E.C. High-Dose Tirilazad for Acute Stroke (RANTTAS II). Stroke 1998, 29. [Google Scholar] [CrossRef] [Green Version]
- Bath, P.M.W.; Blecic, S.; Bogousslavsky, J.; Boysen, G.; Davis, S.; Diez-Tejedor, E.; Ferro, J.M.; Gommans, J.; Hacke, W.; Indredavik, B.; et al. Tirilazad Mesylate in Acute Ischemic Stroke: A systematic review. Tirilazad International Steering Committee. Stroke 2000, 31, 2257–2265. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, S.; Tsuchidate, R.; Smith, M.-L.; Maples, K.R.; Siesjö, B.K. Neuroprotective Effects of a Novel Nitrone, NXY-059, after Transient Focal Cerebral Ischemia in the Rat. J. Cereb. Blood Flow Metab. 1999, 19, 778–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapchak, P.A.; Araujo, D.M.; Song, D.; Wei, J.; Zivin, J.A. Neuroprotective Effects of the Spin Trap Agent Disodium-[(tert-butylimino)methyl]benzene-1,3-disulfonate N-Oxide (Generic NXY-059) in a Rabbit Small Clot Embolic Stroke Model: Combination studies with the thrombolytic tissue plasminogen activator. Stroke 2002, 33, 1411–1415. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.W.; Cummings, R.M.; Bowes, L.J.; Ridley, R.M.; Green, A.R. Functional and Histological Evidence for the Protective Effect of NXY-059 in a Primate Model of Stroke When Given 4 Hours After Occlusion. Stroke 2003, 34, 2228–2233. [Google Scholar] [CrossRef] [Green Version]
- Macleod, M.R.; van der Worp, H.B.; Sena, E.S.; Howells, D.W.; Dirnagl, U.; Donnan, G.A. Evidence for the Efficacy of NXY-059 in Experimental Focal Cerebral Ischaemia Is Confounded by Study Quality. Stroke 2008, 39, 2824–2829. [Google Scholar] [CrossRef] [Green Version]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.-C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U. NXY-059 for the Treatment of Acute Ischemic Stroke. N. Engl. J. Med. 2009, 357, 562–571. [Google Scholar] [CrossRef]
- Savitz, S. A critical appraisal of the NXY-059 neuroprotection studies for acute stroke: A need for more rigorous testing of neuroprotective agents in animal models of stroke. Exp. Neurol. 2007, 205, 20–25. [Google Scholar] [CrossRef]
- Fisher, M.; Lees, K.; Papadakis, M.; Buchan, A.M. NXY-059: Brain or vessel protection. Stroke 2006, 37, 2189–2190. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.; Field, T.; Dowlatshahi, D.; et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Baron, J.-C. Protecting the ischaemic penumbra as an adjunct to thrombectomy for acute stroke. Nat. Rev. Neurol. 2018, 14, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Mayor-Nunez, D.; Ji, Z.; Sun, X.; Teves, L.; Garman, J.D.; Tymianski, M. Plasmin-resistant PSD-95 inhibitors resolve effect-modifying drug-drug interactions between alteplase and nerinetide in acute stroke. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Casals, J.B.; Pieri, N.C.G.; Feitosa, M.L.T.; Ercolin, A.C.M.; Roballo, K.C.S.; Barreto, R.S.N.; Bressan, F.F.; Martins, D.S.; Miglino, M.A.; Ambrósio, C.E. The use of animal models for stroke research: A review. Comp. Med. 2011, 61, 305–313. [Google Scholar]
- Wei, L.; Shi, H.; Lin, X.; Zhang, X.; Wang, Y.; Liu, G.; Xian, X. Impact of Cholesterol on Ischemic Stroke in Different Human-Like Hamster Models: A New Animal Model for Ischemic Stroke Study. Cells 2019, 8, 1028. [Google Scholar] [CrossRef] [Green Version]
- Matter, C.M.; Ma, L.; von Lukowicz, T.; Meier, P.; Lohmann, C.; Zhang, D.; Kilic, U.; Hofmann, E.; Ha, S.-W.; Hersberger, M.; et al. Increased Balloon-Induced Inflammation, Proliferation, and Neointima Formation in Apolipoprotein E (ApoE) Knockout Mice. Stroke 2006, 37, 2625–2632. [Google Scholar] [CrossRef] [Green Version]
- Herz, J.; Sabellek, P.; Lane, T.E.; Gunzer, M.; Hermann, D.M.; Doeppner, T.R. Role of Neutrophils in Exacerbation of Brain Injury After Focal Cerebral Ischemia in Hyperlipidemic Mice. Stroke 2015, 46, 2916–2925. [Google Scholar] [CrossRef] [Green Version]
- Wajngarten, M.; Sempaio, G.; Silva, G.S. Hypertension and Stroke: Update on Treatment. Eur. Cardiol. Rev. 2019, 14, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Möller, K.; Pösel, C.; Kranz, A.; Schulz, I.; Scheibe, J.; Didwischus, N.; Boltze, J.; Weise, G.; Wagner, D.-C. Arterial Hypertension Aggravates Innate Immune Responses after Experimental Stroke. Front. Cell. Neurosci. 2015, 9, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, M.R.; Saad, Y.; Dene, H.; Rapp, J.P. Blood pressure QTL that differentiate Dahl salt-sensitive and spontaneously hypertensive rats. Physiol. Genom. 2000, 3, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhang, Y.; Mo, J.; Su, Z.; Huang, R. Two-Kidney, Two Clip Renovascular Hypertensive Rats Can Be Used as Stroke-prone Rats. Stroke 1998, 29, 1708–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Rehni, A.K.; Liu, A.; Perez-Pinzon, M.A.; Dave, K.R. Diabetic aggravation of stroke and animal models. Exp. Neurol. 2017, 292, 63–79. [Google Scholar] [CrossRef]
- King, A.J. The use of animal models in diabetes research. J. Cereb. Blood Flow Metab. 2012, 166, 877–894. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, S. Impact of obesity-induced type 2 diabetes on long-term outcomes following stroke. Clin. Sci. 2019, 133, 1603–1607. [Google Scholar] [CrossRef]
- Jackman, K.; Kunz, A.; Iadecola, C. Modeling Focal Cerebral Ischemia In Vivo. Methods Mol. Biol. 2011, 793, 195–209. [Google Scholar] [CrossRef]
- Hermann, D.M.; Popa-Wagner, A.; Kleinschnitz, C.; Doeppner, T.R. Animal models of ischemic stroke and their impact on drug discovery. Expert Opin. Drug Discov. 2019, 14, 315–326. [Google Scholar] [CrossRef]
- Tamura, A.; Graham, D.I.; McCulloch, J.; Teasdale, G.M. Focal Cerebral Ischaemia in the Rat: 1. Description of Technique and Early Neuropathological Consequences following Middle Cerebral Artery Occlusion. J. Cereb. Blood Flow Metab. 1981, 1, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, W.D.; Watson, B.D.; Busto, R.; Ginsberg, M.D.; Bethea, J.R. Photochemically induced cerebral infarction. I. Early microvascular alterations. Acta Neuropathol. 1987, 72, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Fluri, F.; Schuhmann, M. Animal models of ischemic stroke and their application in clinical research. Drug Des. Dev. Ther. 2015, ume 9, 3445–3454. [Google Scholar] [CrossRef] [Green Version]
- Biernaskie, J.; Corbett, D.; Peeling, J.; Wells, J.; Lei, H. A serial MR study of cerebral blood flow changes and lesion development following endothelin-1-induced ischemia in rats. Magn. Reson. Med. 2001, 46, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, M.; Kasuya, Y.; Hayashi, K.; Goto, K. SB209670, a potent endothelin receptor antagonist, prevents or delays axonal degeneration after spinal cord injury. Brain Res. 1998, 786, 235–239. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Technique | Procedure | Advantage | Disadvantage |
|---|---|---|---|
| Middle cerebral artery occlusion |
|
|
|
| Transcranial occlusion |
|
|
|
| Cerebral photothrombosis |
|
|
|
| Endothelin-1 occlusion |
|
|
|
| Cerebral embolism |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haupt, M.; Gerner, S.T.; Bähr, M.; Doeppner, T.R. Quest for Quality in Translational Stroke Research—A New Dawn for Neuroprotection? Int. J. Mol. Sci. 2022, 23, 5381. https://doi.org/10.3390/ijms23105381
Haupt M, Gerner ST, Bähr M, Doeppner TR. Quest for Quality in Translational Stroke Research—A New Dawn for Neuroprotection? International Journal of Molecular Sciences. 2022; 23(10):5381. https://doi.org/10.3390/ijms23105381
Chicago/Turabian StyleHaupt, Matteo, Stefan T. Gerner, Mathias Bähr, and Thorsten R. Doeppner. 2022. "Quest for Quality in Translational Stroke Research—A New Dawn for Neuroprotection?" International Journal of Molecular Sciences 23, no. 10: 5381. https://doi.org/10.3390/ijms23105381
APA StyleHaupt, M., Gerner, S. T., Bähr, M., & Doeppner, T. R. (2022). Quest for Quality in Translational Stroke Research—A New Dawn for Neuroprotection? International Journal of Molecular Sciences, 23(10), 5381. https://doi.org/10.3390/ijms23105381