
Comparative Analyses of Medicinal Chemistry and Cheminformatics Filters with Accessible Implementation in Konstanz Information Miner (KNIME)



Abstract
:1. Introduction
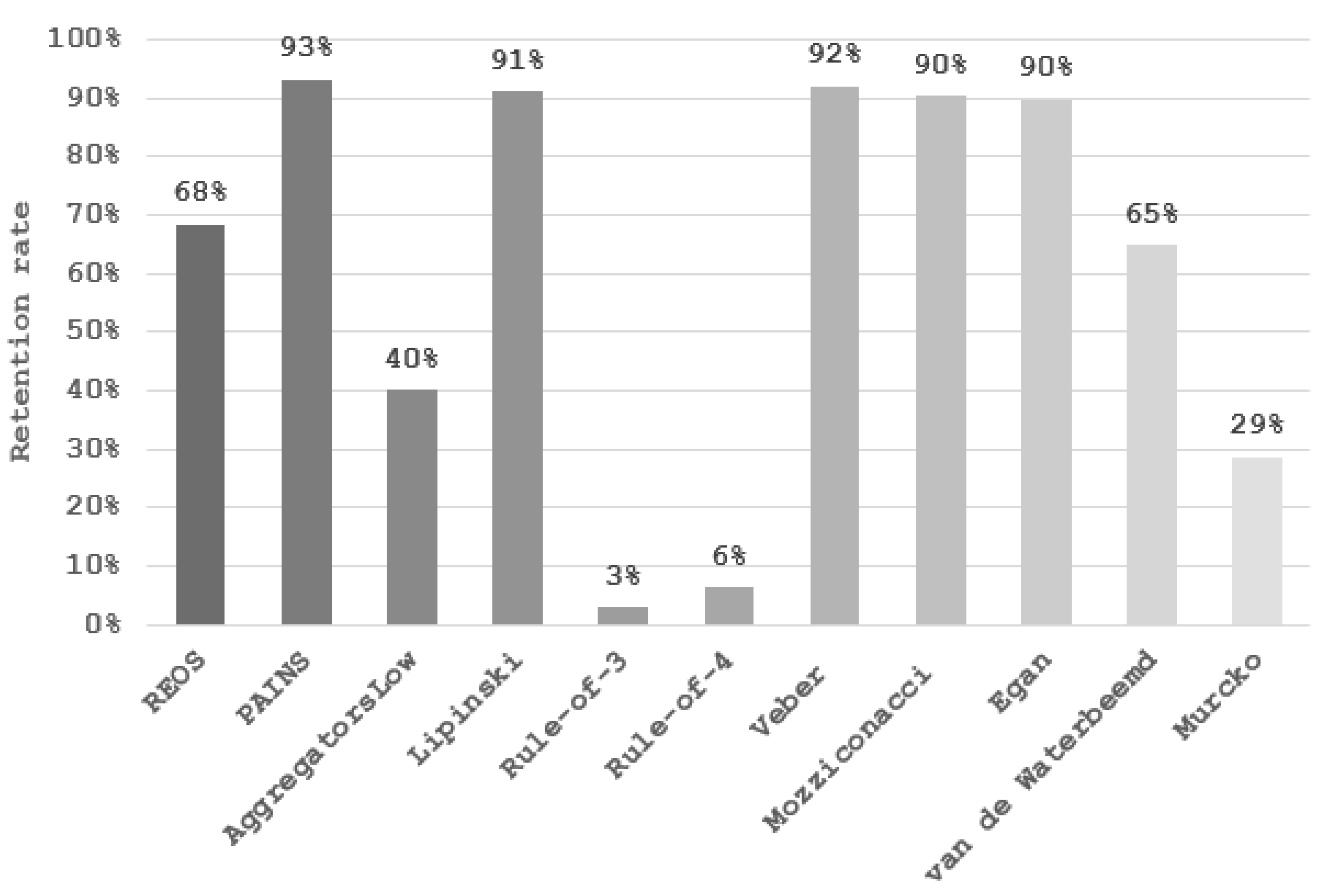
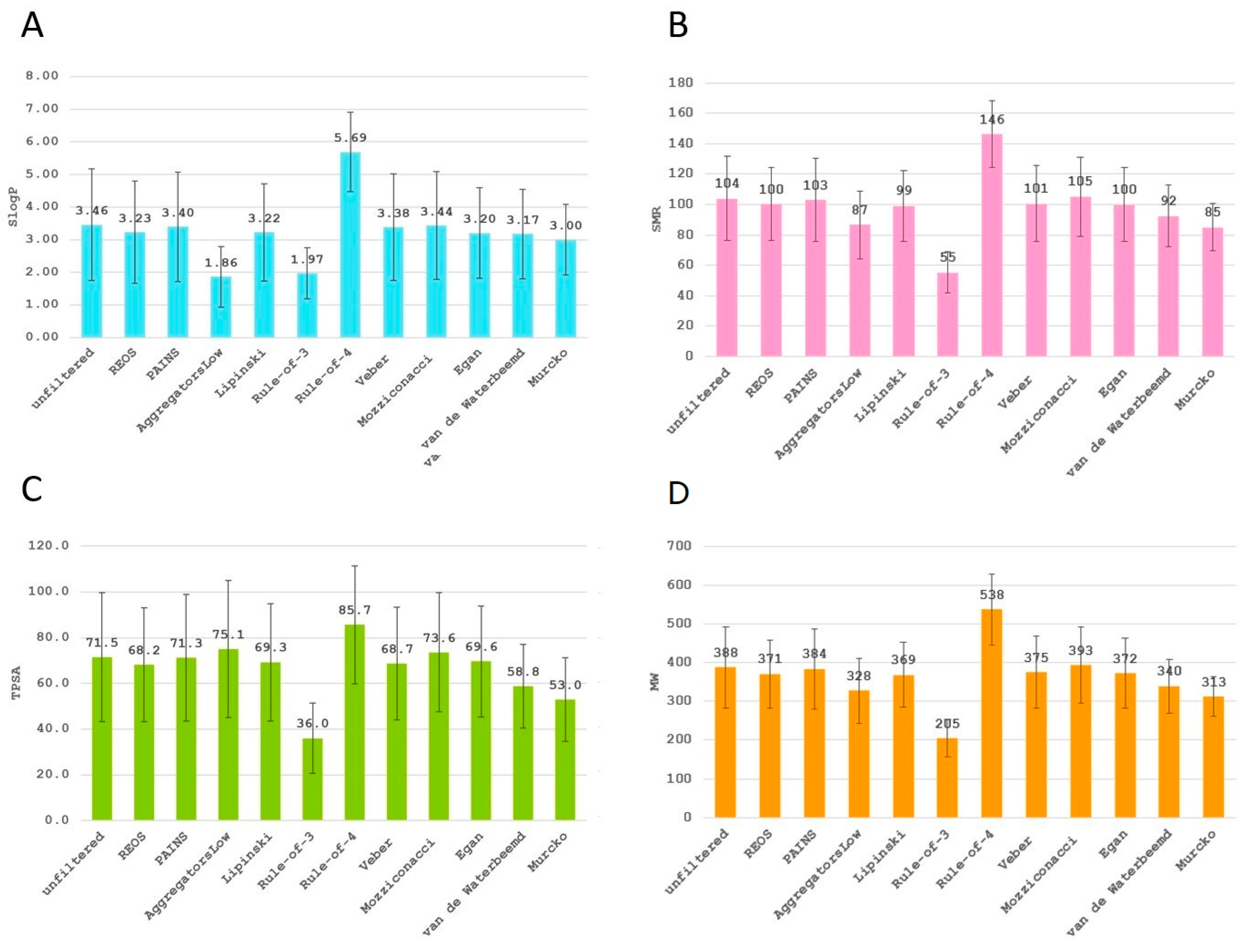
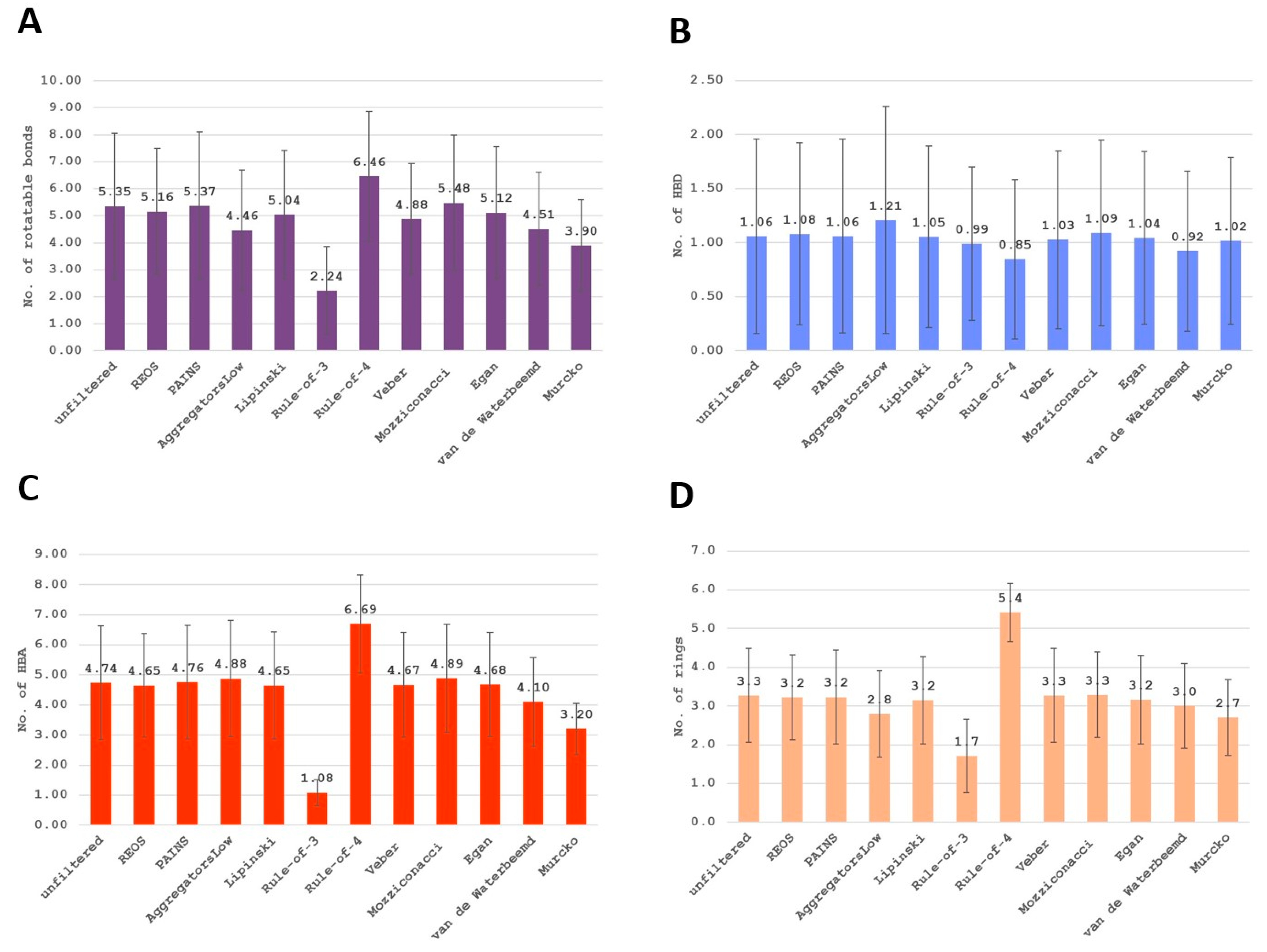
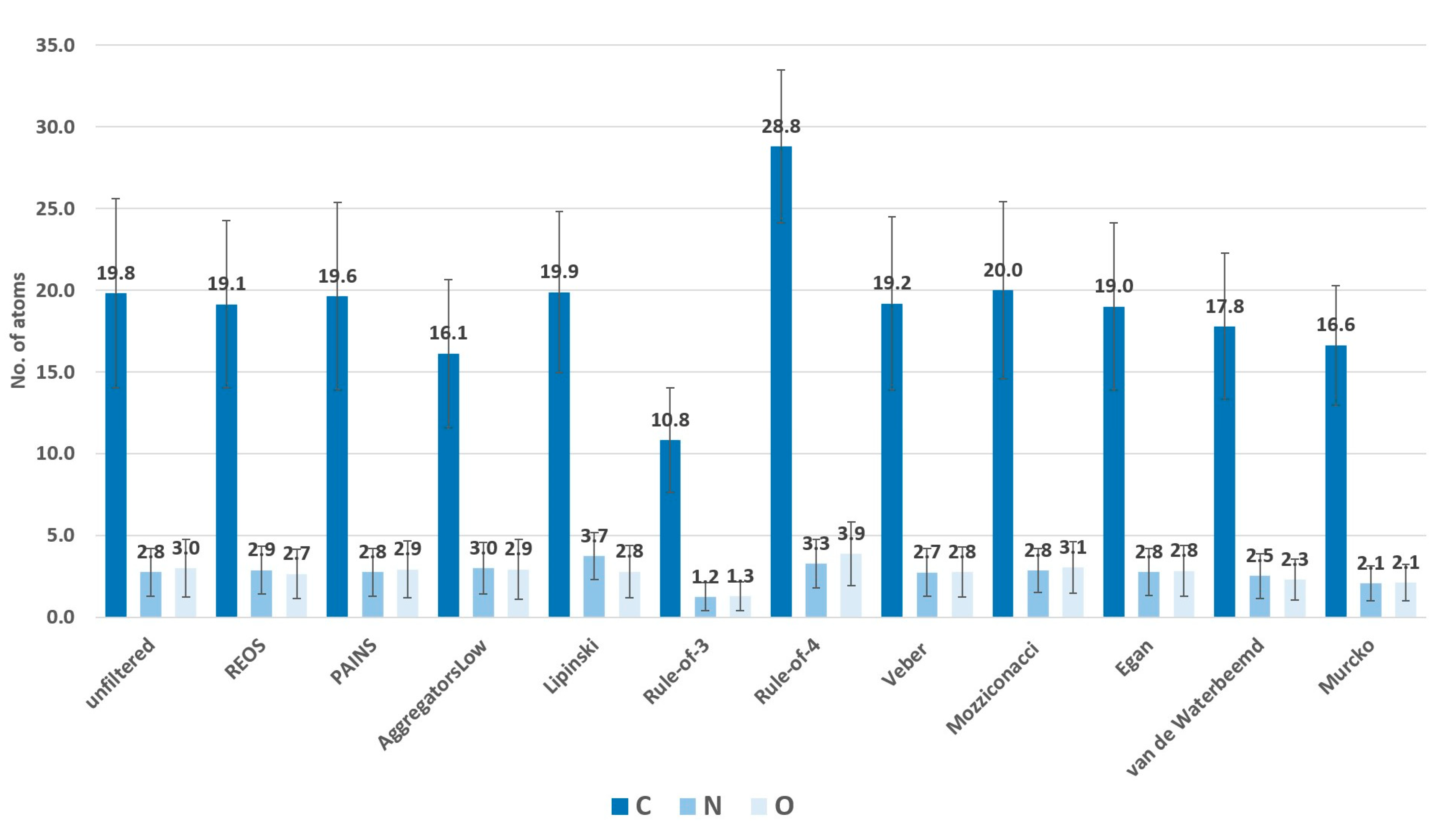
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CC | combinatorial chemistry |
| HTS | high-throughput screening |
| VS | virtual screening |
| HTVS | high-throughput virtual screening |
| ADME | absorption, distribution, metabolism, and excretion |
| MW | molecular weight |
| Ro4 | Rule-of-4 |
| Ro5 | Rule-of-5 |
| Ro3 | Rule-of-3 |
| SDF | structure-data file format |
| MOL | MDL Molfile |
| SMILES | simplified molecular input line entry specification format |
| SMARTS | SMILES arbitrary target specification |
| KNIME | Konstanz Information Miner |
| SD | standard deviations |
| SMR | molecular refractivity |
| TPSA | total polar surface area |
| MW | molecular weight |
| HBA | No. of hydrogen bond acceptors |
| HBD | No. of hydrogen bond donors |
| REOS | rapid elimination of swill |
| PAINS | pan-assay interference compounds |
| CNS | central nervous system |
References
- Oprea, T.I. Property Distribution of Drug-Related Chemical Databases. J. Comput. Aided Mol. Des. 2000, 14, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial Chemistry in Drug Discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P.; Murcko, M.A. Prediction of “Drug-Likeness”. Adv. Drug Deliv. Rev. 2002, 54, 255–271. [Google Scholar] [CrossRef]
- Bakken, G.A.; Bell, A.S.; Boehm, M.; Everett, J.R.; Gonzales, R.; Hepworth, D.; Klug-McLeod, J.L.; Lanfear, J.; Loesel, J.; Mathias, J.; et al. Shaping a Screening File for Maximal Lead Discovery Efficiency and Effectiveness: Elimination of Molecular Redundancy. J. Chem. Inf. Model. 2012, 52, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Njoroge, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.B.; Smith, P.W.; Chibale, K. Recent Approaches to Chemical Discovery and Development against Malaria and the Neglected Tropical Diseases Human African Trypanosomiasis and Schistosomiasis. Chem. Rev. 2014, 114, 11138–11163. [Google Scholar] [CrossRef]
- Morgan, P.; Brown, D.G.; Lennard, S.; Anderton, M.J.; Barrett, J.C.; Eriksson, U.; Fidock, M.; Hamrén, B.; Johnson, A.; March, R.E.; et al. Impact of a Five-Dimensional Framework on R&D Productivity at AstraZeneca. Nat. Rev. Drug Discov. 2018, 17, 167–181. [Google Scholar] [CrossRef]
- Náray-Szabó, G. Analysis of Molecular Recognition: Steric Electrostatic and Hydrophobic Complementarity. J. Mol. Recognit. 1993, 6, 205–210. [Google Scholar] [CrossRef]
- Walters, W.P.; Stahl, M.T.; Murcko, M.A. Virtual Screening—An Overview. Drug Discov. Today 1998, 3, 160–178. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Galloway, W.R.J.D.; Spring, D.R. A Question of Library Design. Nature 2011, 470, 42–43. [Google Scholar] [CrossRef]
- Kralj, S.; Jukič, M.; Bren, U. Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein–Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design. IJMS 2021, 23, 393. [Google Scholar] [CrossRef]
- Macarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.S.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of High-Throughput Screening in Biomedical Research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, D.S.; Edith Chan, A.W.; Binnie, A.; Chen, L.C.; Robinson, A.; Spoonamore, J.; Rodwell, D.; Wade, S.; Wilson, S.; Ackerman-Berrier, M.; et al. Efficient Discovery of Inhibitory Ligands for Diverse Targets from a Small Combinatorial Chemical Library of Chimeric Molecules. Biochem. Biophys. Res. Commun. 1999, 266, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- van De Waterbeemd, H.; Camenisch, G.; Folkers, G.; Raevsky, O.A. Estimation of Caco-2 Cell Permeability Using Calculated Molecular Descriptors. Quant. Struct.-Act. Relat. 1996, 15, 480–490. [Google Scholar] [CrossRef]
- Oprea, T. Virtual Screening in Lead Discovery: A Viewpoint. Molecules 2002, 7, 51–62. [Google Scholar] [CrossRef]
- Walters, W.P.; Murcko, A.A.; Murcko, M.A. Recognizing Molecules with Drug-like Properties. Curr. Opin. Chem. Biol. 1999, 3, 384–387. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Muegge, I. Pharmacophore Features of Potential Drugs. Chemistry 2002, 8, 1976–1981. [Google Scholar] [CrossRef]
- Morelli, X.; Bourgeas, R.; Roche, P. Chemical and Structural Lessons from Recent Successes in Protein–Protein Interaction Inhibition (2P2I). Curr. Opin. Chem. Biol. 2011, 15, 475–481. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the Chemical Beauty of Drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Charifson, P.S.; Walters, W.P. Filtering Databases and Chemical Libraries. J. Comput. Aided Mol. Des. 2002, 16, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Lumley, J.A. Compound Selection and Filtering in Library Design. QSAR Comb. Sci. 2005, 24, 1066–1075. [Google Scholar] [CrossRef]
- Senger, M.R.; Fraga, C.A.M.; Dantas, R.F.; Silva, F.P. Filtering Promiscuous Compounds in Early Drug Discovery: Is It a Good Idea? Drug Discov. Today 2016, 21, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Davis, A.M.; Teague, S.J.; Leeson, P.D. Is There a Difference between Leads and Drugs? A Historical Perspective. J. Chem. Inf. Comput. Sci. 2001, 41, 1308–1315. [Google Scholar] [CrossRef]
- Olah, M.M.; Bologa, C.G.; Oprea, T.I. Strategies for Compound Selection. Curr. Drug Discov. Technol. 2004, 1, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Lenci, E.; Trabocchi, A. Peptidomimetic Toolbox for Drug Discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef]
- Jukič, M.; Janežič, D.; Bren, U. Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors. Molecules 2020, 25, 5808. [Google Scholar] [CrossRef]
- Dalby, A.; Nourse, J.G.; Hounshell, W.D.; Gushurst, A.K.I.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of Several Chemical Structure File Formats Used by Computer Programs Developed at Molecular Design Limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255. [Google Scholar] [CrossRef]
- Hähnke, V.D.; Kim, S.; Bolton, E.E. PubChem Chemical Structure Standardization. J. Cheminform. 2018, 10, 36. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 2015, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Cao, S.; Su, P.-C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit Identification and Optimization in Virtual Screening: Practical Recommendations Based on a Critical Literature Analysis: Miniperspective. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajay; Bemis, G.W.; Murcko, M.A. Designing Libraries with CNS Activity. J. Med. Chem. 1999, 42, 4942–4951. [Google Scholar] [CrossRef] [PubMed]
- Thorne, N.; Auld, D.S.; Inglese, J. Apparent Activity in High-Throughput Screening: Origins of Compound-Dependent Assay Interference. Curr. Opin. Chem. Biol. 2010, 14, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rishton, G.M. Nonleadlikeness and Leadlikeness in Biochemical Screening. Drug Discov. Today 2003, 8, 86–96. [Google Scholar] [CrossRef]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58, 7076–7087. [Google Scholar] [CrossRef] [Green Version]
- Bruns, R.F.; Watson, I.A. Rules for Identifying Potentially Reactive or Promiscuous Compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Walters, W.P.; Namchuk, M. Designing Screens: How to Make Your Hits a Hit. Nat. Rev. Drug Discov. 2003, 2, 259–266. [Google Scholar] [CrossRef]
- Bush, B.L.; Sheridan, R.P. PATTY: A Programmable Atom Type and Language for Automatic Classification of Atoms in Molecular Databases. J. Chem. Inf. Comput. Sci. 1993, 33, 756–762. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Fichert, T.; Yazdanian, M.; Proudfoot, J.R. A Structure-Permeability Study of Small Drug-like Molecules. Bioorg. Med. Chem. Lett. 2003, 13, 719–722. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.L.; Schneider, G. Scaffold Architecture and Pharmacophoric Properties of Natural Products and Trade Drugs: Application in the Design of Natural Product-Based Combinatorial Libraries. J. Comb. Chem. 2001, 3, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Mozziconacci, J.C.; Arnoult, E.; Baurin, N.; Marot, C. Preparation of a Molecular Database from a Set of 2 Million Compounds for Virtual Screening Applications: Gathering, Structural Analysis and Filtering. In Proceedings of the 9th Electronic Computational Chemistry Conference, World Wide Web, 1–31 March 2003. [Google Scholar]
- Darvas, F.; Keseru, G.; Papp, A.; Dorman, G.; Urge, L.; Krajcsi, P. In Silico and Ex Silico ADME Approaches for Drug Discovery. CTMC 2002, 2, 1287–1304. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A “rule of Three” for Fragment-Based Lead Discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- van de Waterbeemd, H.; Camenisch, G.; Folkers, G.; Chretien, J.R.; Raevsky, O.A. Estimation of Blood-Brain Barrier Crossing of Drugs Using Molecular Size and Shape, and H-Bonding Descriptors. J. Drug Target. 1998, 6, 151–165. [Google Scholar] [CrossRef]
- van de Waterbeemd, H. Physicochemical Approaches to Drug Absorption. In Methods and Principles in Medicinal Chemistry; van de Waterbeemd, H., Testa, B., Eds.; Wiley: Hoboken, NJ, USA, 2008; Volume 40, pp. 69–99. ISBN 978-3-527-32051-6. [Google Scholar]
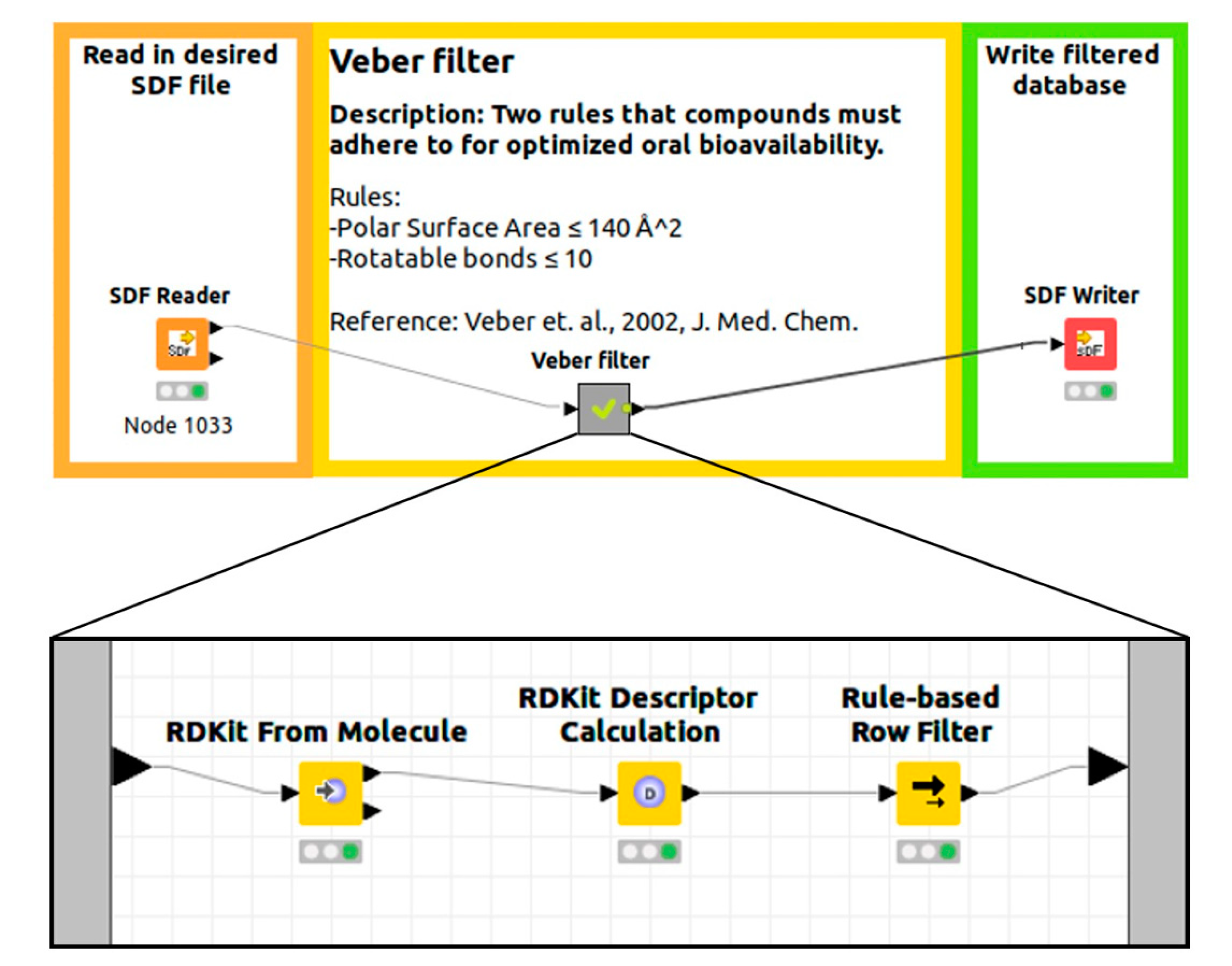
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name/Reference | Description | Features/Cutoff Values |
|---|---|---|
| Aggregators [36] | Tanimoto coefficient similarity search to a database of known aggregators. | Tanimoto coefficient similarity ≥ 0.85 or SlogP > 5 (high similarity), |
| Tanimoto coefficient similarity ≥ 0.5 and SlogP > 3 (medium similarity), | ||
| Tanimoto coefficient similarity < 0.85 and SlogP ≤ 3 (low similarity) | ||
| Ely Lilly Rules [37] | A set of 275 rules, developed over an 18-year period, used to identify compounds that may interfere with biological assays, allowing their removal from screening sets. | Reasons for rejection of compounds: reactivity, interference with assay measurements (fluorescence, absorbance, quenching), instability and lack of druggability (lacking both oxygen and nitrogen) |
| Muegge method [18,38] | Bioavailability prediction rules dubbed the Muegge method. Pharmacophore filter developed by analyzing known drug databases, with four functional molecular motifs determined to be important in drug-like molecules: | Primary, secondary, and tertiary amines are considered pharmacophore points but not pyrrole, indole, thiazole, isoxazole, other azoles, or diazines. Compounds with more than one carboxylic acid are dismissed. Compounds without a ring structure are dismissed. Intracyclic amines that occur in the same ring are fused and count as only one pharmacophore point. |
| PAINS [39] | Removal of frequent hitters (promiscuous compounds) by identifying sub-structural features not recognized by filters commonly used to identify reactive compounds. | Functional groups such as rhodanines, phenolic Mannich bases, hydroxyphenylhydrazones, alkylidene barbiturates, alkylidene heterocycles, 1,2,3-aralkylpyrroles, activated benzofurazans, 2-amino-3-carbonylthiophenes, catechols, and quinones do not pass the filters. |
| REOS 1 [3,40,41] | Seven property filters | H-bond donor ≤ 5, |
| (similar to the PATTY | H-bond acceptors ≤ 10, | |
| rules in program developed at Merck) | −2 ≤ Formal charge ≤ +2, | |
| Number of rotatable bonds ≤ 8, | ||
| 200 ≤ Molecular weight ≤ 500, | ||
| 20 ≤ number of heavy atoms ≤ 50, | ||
| −2 ≤ logP ≤ 5 | ||
| Functional group filters for the removal of problematic structures dubbed REOS (rapid elimination of swill; program developed at Vertex). | Reactive, toxic and other undesirable moieties such as nitro groups, preoxides, triflates, aldehydes, acetals, etc. |
| Name/Reference | Description | Features/Cutoff Values |
|---|---|---|
| Egan [42] | Set of rules designed by analyzing the data on compounds both well and poorly absorbed in humans with multivariate statistics. Two descriptors (AlogP and PSA) were chosen for inclusion when determining membrane permeability. Compounds that pass exhibit good bioavailability. | AlogP ≤ 5.88, |
| polar surface area ≤ 131.6 Å2 | ||
| Fichert [43] | Rules for structure-permeability based on a set of 41 small drug-like molecules. LogD is the main property that determines permeability, with structures passing this filter being highly permeable in the Cacao-2 model. | Molecular weight ≤ 500, |
| 0 ≤ logD ≤ 3 | ||
| Ghose [44] | A set of rules for drug-likeness derived from characterizing 6304 compounds taken from the Comprehensive Medicinal Chemistry Database. | 180 ≤ molecular weight ≤ 480, |
| 40 ≤ molecular refractivity ≤ 130, | ||
| −0.4 ≤ ClogP ≤ 5.6, | ||
| 20 ≤ number of atoms ≤ 70 | ||
| Lee filter [45] | Analysis of natural products to determine potential appealing scaffolds for future drug design. Pharmacophoric properties of natural products, trade drugs, and virtual combinatorial library were assessed, finding key properties and several scaffolds which could work as building blocks. | MW mean ~356 |
| LogP mean ~2.1 | ||
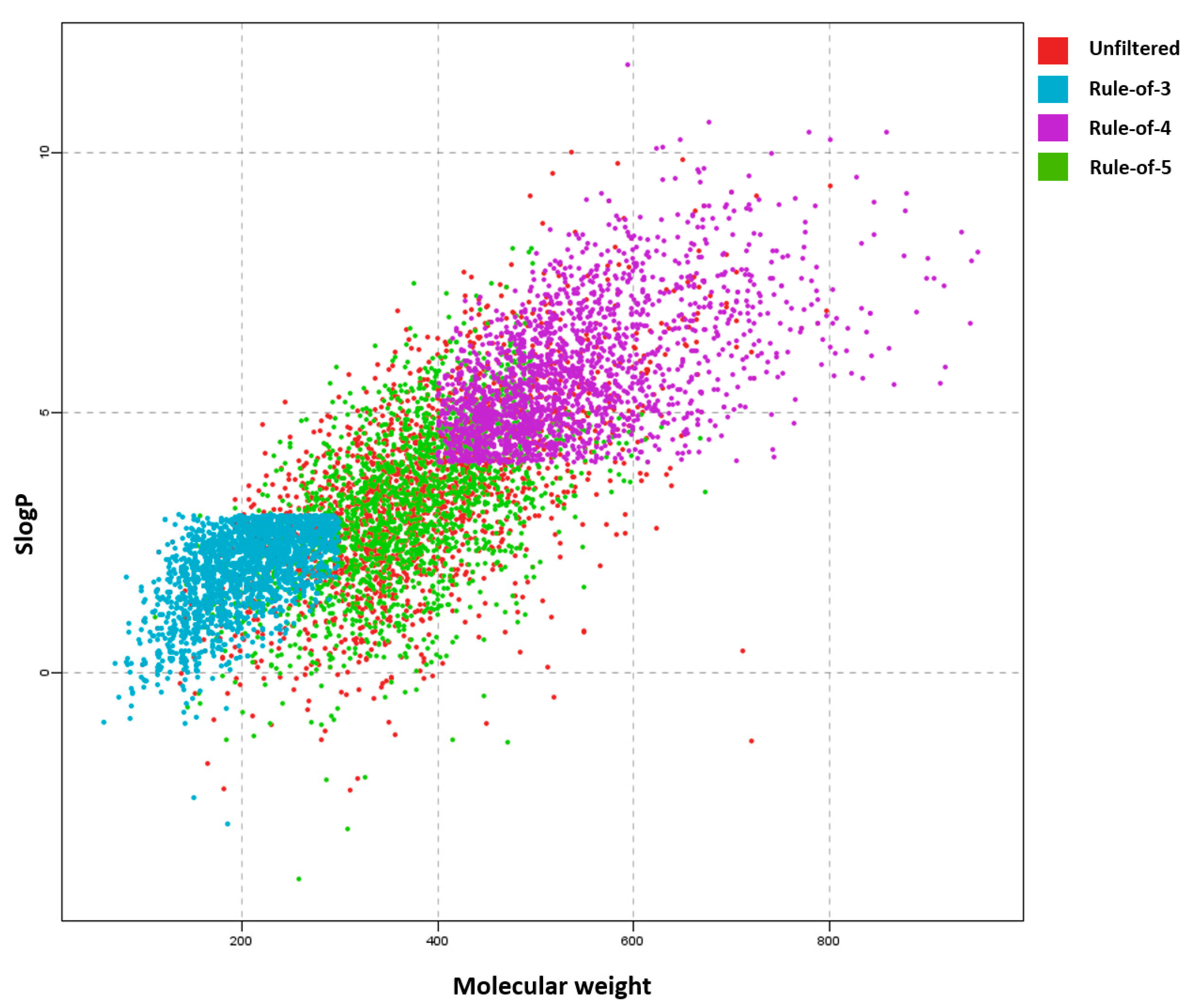
| Lipinski (Rule-of-5) [13] | A set of four rules for drug-likeness and oral bioavailability derived from a subset of 2245 drugs from the World Drug Index. The rules aim to address the ADME issues. | Molecular weight ≤ 500, |
| logP ≤ 5, | ||
| H-bond donors ≤ 5, | ||
| H-bond acceptors ≤ 10 | ||
| Mozzicconacci [46] | Filter developed by Mozziconacci after analyzing 15 freely available chemical libraries (2 million compounds). Drug-likeness was examined using common chemical features and based on the successive filters were designed to extract the drug-like subset. | Rotatable bonds ≤ 15, |
| number of rings ≤ 6, | ||
| oxygen atoms ≥ 1, | ||
| nitrogen atoms ≥ 1, | ||
| halogen atoms ≤ 7 | ||
| Murcko filter [33,47] | Rules for determining CNS activity, joining 7 property descriptors (Rule-of-5 with the addition of rotatable bonds, aromatic density, and a measure for branching) and 166 fingerprint descriptors to determine presence or absence of functional groups. | MW 200–540, |
| logP 0–5.2, | ||
| H-bond acceptors ≤ 4, | ||
| H-bond donor ≤ 3, | ||
| rotatable bonds ≤ 7, | ||
| branching behavior 3.4–12.2, | ||
| aromatic rings < 3 | ||
| Oprea Lead-Like [1,24] | A set of rules based on lead-like vs. drug-like comparison after examination of several commercially available databases. The rules aim to maintain focus towards effective and orally absorbable compounds. Beside the properties chosen based on the Rule-of-5, additional properties were chosen to better reflect molecular complexity of a library and the rigidity of a molecule. | Molecular weight < 450, |
| −3.5 ≤ logP < 4.5, | ||
| −4 ≤ logD ≤ 4, | ||
| number of rings ≤ 4, | ||
| nonterminal single bonds ≤ 10, | ||
| H-bond donor ≤ 5, | ||
| H-bond acceptor ≤ 8 | ||
| Rule-of-3 [48] | Rules designed to support “fragment-based” drug research. Hits obtained using this filter can be useful for fragment libraries used to generate potential leads. Fragment libraries are useful for sampling chemical diversity or targeting specific interactions. | Molecular weight ≤ 300, |
| logP ≤ 3, | ||
| H-bond donor ≤ 3, | ||
| H-bond acceptors ≤ 3 | ||
| rotatable bonds ≤ 3 | ||
| Rule-of-4 [19] | A set of rules derived from analyzing the 2P2I database that contains protein–protein interaction inhibitors with the aim of establishing guidelines for druggable protein–protein inhibitors, since these most often break traditional property filter rules. | Molecular weight ≥ 400, |
| logP ≥ 4, | ||
| number of rings ≥ 4, | ||
| H-bond acceptors ≥ 4 | ||
| van de Waterbeemd [49,50] | Physiochemical properties for estimation of blood–brain barrier crossing of compounds. Rules were derived by examination of lipophilicity, H-bonding capacity, and molecular shape and size descriptors of marketed CNS and CNS-inactive drugs. | Molecular weight ≤ 450, |
| polar surface area ≤ 90 Å2 | ||
| Veber [51] | Two rules to meet the criteria for oral bioavailability derived after studying bioavailability measurements in rats for of over 1100 drug candidates at GlaxoSmithKline. | Rotatable bonds ≤ 10, |
| polar surface area ≤ 140 Å2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kralj, S.; Jukič, M.; Bren, U. Comparative Analyses of Medicinal Chemistry and Cheminformatics Filters with Accessible Implementation in Konstanz Information Miner (KNIME). Int. J. Mol. Sci. 2022, 23, 5727. https://doi.org/10.3390/ijms23105727
Kralj S, Jukič M, Bren U. Comparative Analyses of Medicinal Chemistry and Cheminformatics Filters with Accessible Implementation in Konstanz Information Miner (KNIME). International Journal of Molecular Sciences. 2022; 23(10):5727. https://doi.org/10.3390/ijms23105727
Chicago/Turabian StyleKralj, Sebastjan, Marko Jukič, and Urban Bren. 2022. "Comparative Analyses of Medicinal Chemistry and Cheminformatics Filters with Accessible Implementation in Konstanz Information Miner (KNIME)" International Journal of Molecular Sciences 23, no. 10: 5727. https://doi.org/10.3390/ijms23105727
APA StyleKralj, S., Jukič, M., & Bren, U. (2022). Comparative Analyses of Medicinal Chemistry and Cheminformatics Filters with Accessible Implementation in Konstanz Information Miner (KNIME). International Journal of Molecular Sciences, 23(10), 5727. https://doi.org/10.3390/ijms23105727