1. Introduction
Antibody phage display is utilized widely for the development of monoclonal antibodies, due in large part to the in vitro nature of the technology, which enables the isolation of human antibodies with exquisite antigen specificities [
1]. Many therapeutic antibodies have been discovered or optimized by phage display technology [
2], validating its utility as a technological platform for biopharmaceutical drug development. Antibody libraries with high diversity and sophisticated designs have been constructed and shown to produce high-quality antibodies with desired functional activities suitable for therapeutic applications [
3,
4,
5,
6,
7,
8,
9,
10].
Despite its apparent technological advantages, however, antibody phage display has not entirely displaced antibody discovery from the immunized animal; on the contrary, animal-derived monoclonal antibodies (including chimeric, humanized, and fully human antibodies) constitute a majority of therapeutic antibodies used currently in clinic or under clinical evaluation [
11]. It has been suggested that the eukaryotic quality control machineries may favor antibody clones with better physicochemical properties and expression efficiency [
12], critical attributes for the successful development of therapeutic antibodies. Another factor that distinguishes the in vivo antibody generation process from the in vitro phage display approach is the linkage between binding and amplification. B cell activation and proliferation require antigen binding to a functional B cell receptor (BCR) and the subsequent signaling events, and the absence of in-frame BCR or the lack of antigen binding to BCR results in B cell apoptosis [
13]. In theory, a similar mechanism is applied to antibody phage display; antigen-binding phage clones are selected and amplified in
Escherichia coli host cells, whereas non-binders are washed out and discarded. However, unlike the B cell activation by BCR signaling, the binding and amplification steps of phage display are separated spatially and temporally. As a result, a large number of nonbinding clones are retrieved and amplified, and, because some of them—especially non-producing clones with early stop codons—have a sufficient growth advantage to compensate for the loss in numbers during wash cycles, they may overwhelm the output after multiple rounds of biopanning.
Rapid amplification of nonbinding, fast-growing clones can be minimized by a number of strategies, most notably the engineered helper phages lacking a functional copy of
gIII [
14,
15,
16,
17,
18]. Since infection of
Escherichia coli by M13 bacteriophage requires the minor coat protein pIII, phages rescued by such an engineered helper phage cannot infect the host unless it displays a foreign protein fused to a functional, phagemid-derived pIII. Although this is an effective strategy for excluding non-producing clones, it cannot prevent the amplification of highly productive nonbinders; furthermore, such characteristics as the multivalent display, low production titer, or incompatibility with the common phage display features (e.g., the use of a phagemid with truncated
gIII or an amber stop codon) may limit wider applicability of this strategy [
15]. Another, probably more fundamental, solution to the problem is to design and construct a library consisting of antibody clones with desirable physicochemical profiles. For example, hundreds of human VH/VL pairs were screened for stability, expression, display level, and monomeric contents in order to design ideal frameworks for a synthetic human Fab library [
9].
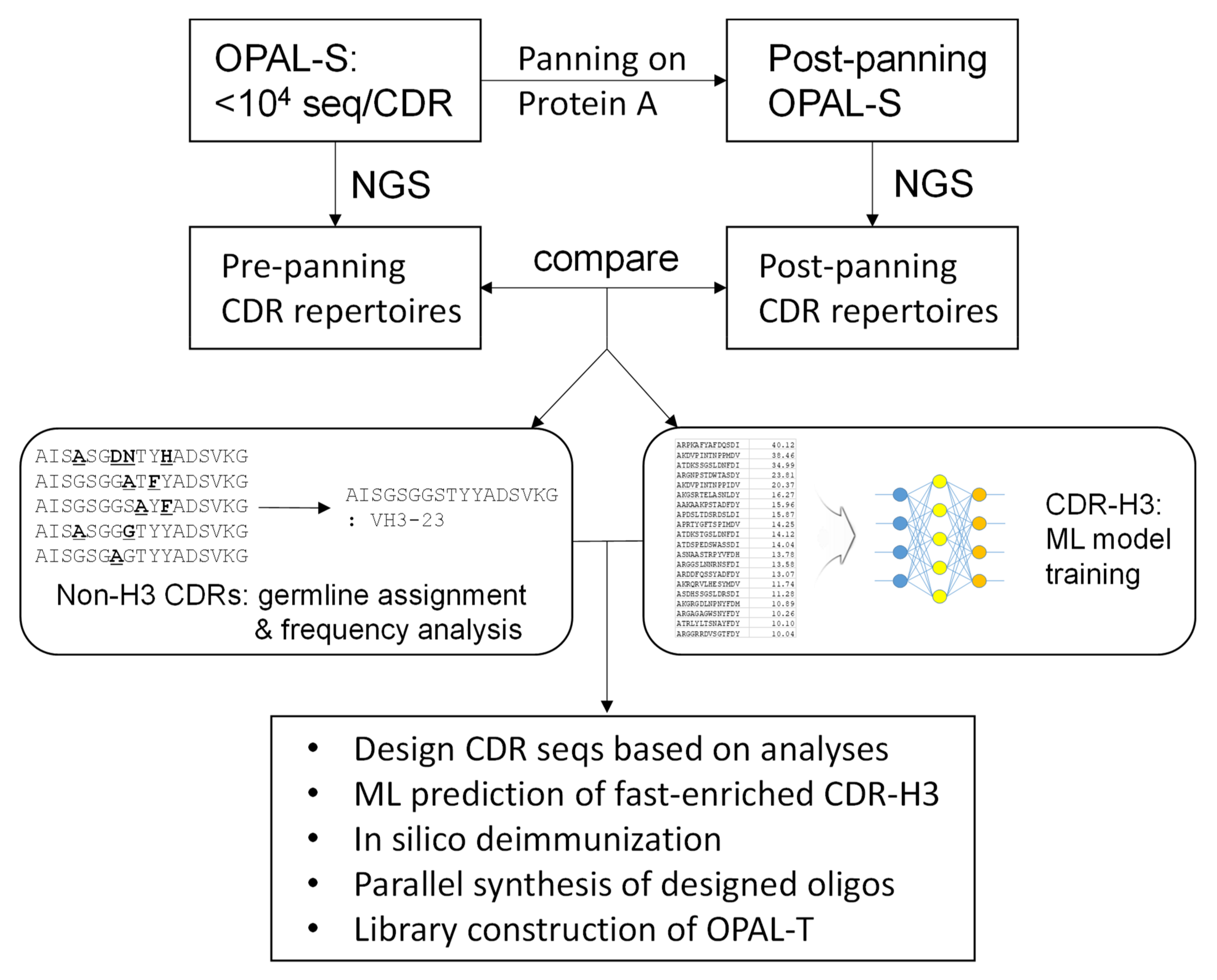
In this report, we attempted to alleviate the problem of amplification bias by designing an antibody library consisting of clones with a better amplification profile and that are thus less likely to be outcompeted by non-binders during the amplification phase of biopanning (
Figure 1). Using a previously constructed library with low, non-combinatorial complementarity-determining region (CDR) diversities (OPAL-S) [
3], single-chain variable fragment (scFv) clones that were amplified and/or displayed more efficiently were enriched by panning against protein A from
Staphylocucccus aureus. Protein A is primarily known for its binding activity to the Fc portion of immunoglobulins [
19], but it also binds to variable domains belonging to human VH3 subgroup [
20]; this unique characteristic has been utilized to select well-folded and highly displayed antibody fragments from phage libraries [
10,
21,
22]. The effect of CDR sequences on the panning enrichment of antibody clones was evaluated by comparing the next generation sequencing (NGS) repertoires of the library before and after panning. CDR sequences for the new library were designed based on the NGS analysis and, especially for CDR-H3, machine learning (ML) models were trained from the sequencing data and utilized to predict fast-amplified sequences. The new library, OPAL-T, generally performed better than OPAL-S when tested against a panel of antigens, highlighting the effectiveness of the antibody library design strategy focused on amplification efficiency.
3. Discussion
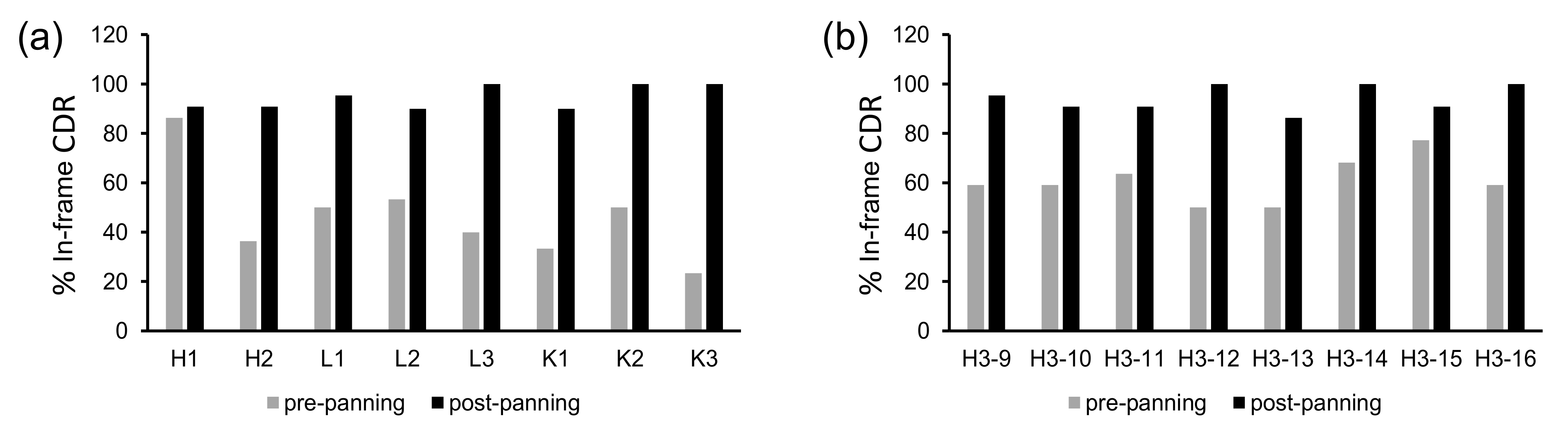
During panning of a phage-displayed antibody library, the selection outcome is determined not only by the binding affinity to the target antigen, but also in large part by the display level and/or growth rates of the clones. The diversity of the panning output may be reduced greatly as a result, because of the preferential enrichment of a small number of binders with significant display/growth advantage, or the faster amplification of some non-binders or non-producers. Even within a synthetic antibody library that employs a fixed framework scaffold, wide variation in the amplification efficiency among the clones was observed, suggesting that CDR sequences have a significant influence on the enrichment of antibody clones during biopanning.
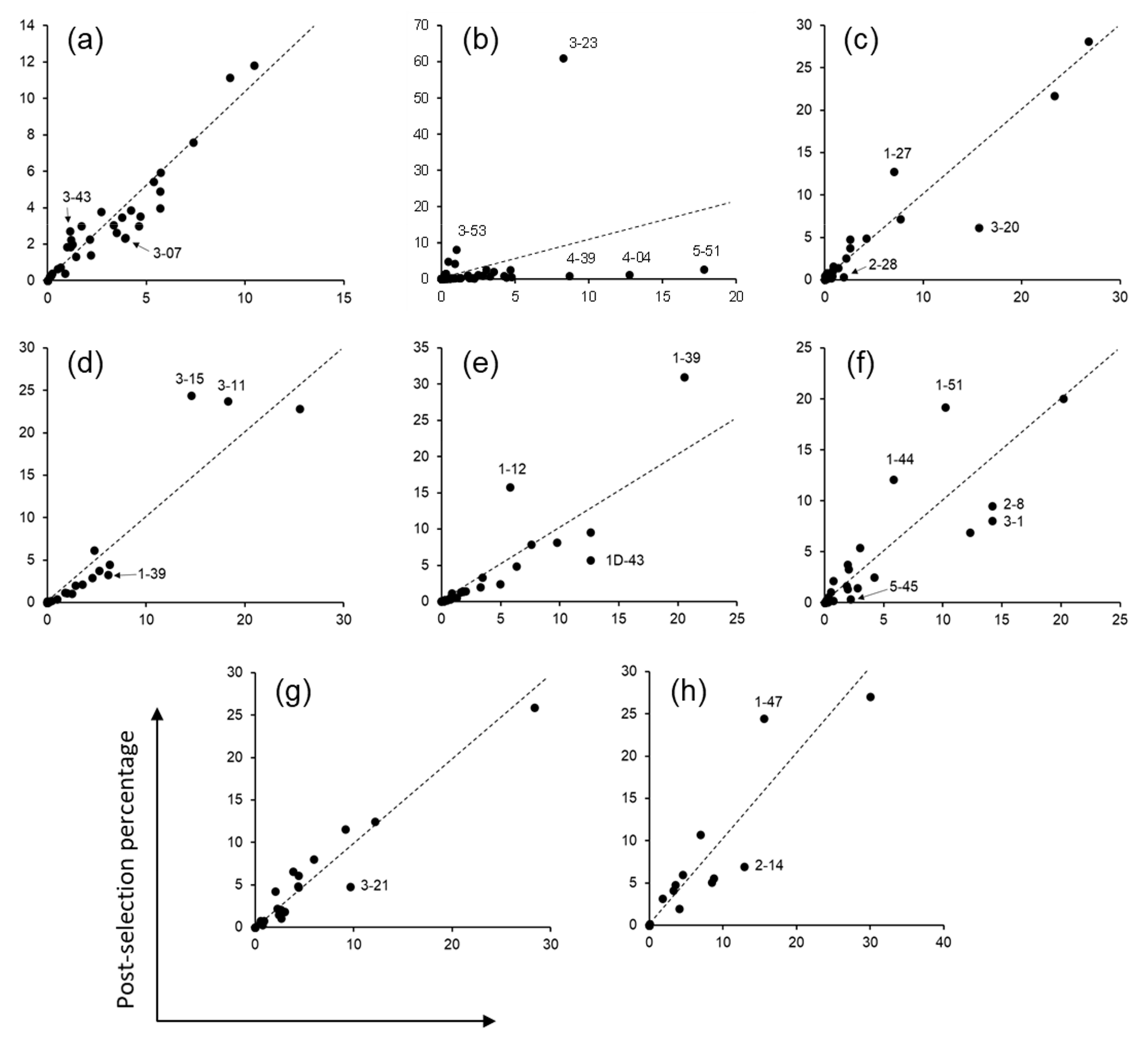
A synthetic scFv library with pre-defined CDR sequences was designed and constructed previously (OPAL-S) [
3]. It should be noted that although the reported size of OPAL-S was 8 × 10
8, the library used in this study was expanded to 1.3 × 10
10 through additional transformations and is comparable in size to OPAL-T (1.4 × 10
10, Table 1). Since the designed diversity of each CDR in this library was between 10
3–10
4, we were able to track the change in relative abundance of every predefined CDR sequence before and after biopanning against protein A by NGS. Non-H3 CDR sequences of OPAL-S were designed individually by simulating somatic hypermutations to the human germline CDR sequences, and thus they were traceable to their germline ancestors. From the analysis of panning enrichment patterns of the CDR sequences, with the exception of CDR-H2, the germline origin of CDR sequences appeared not to have marked influence on the amplification of scFv clones in the context of the VH3-23, Vκ3-20, and Vλ1-47 scaffolds. This conclusion does not necessarily mean that a specific CDR sequence has no influence on the amplification of the scFv; rather, it suggests that no major intrinsic incompatibility exists between those scaffold sequences and the CDRs from different germline genes or families, in terms of the levels of phage display and amplification.
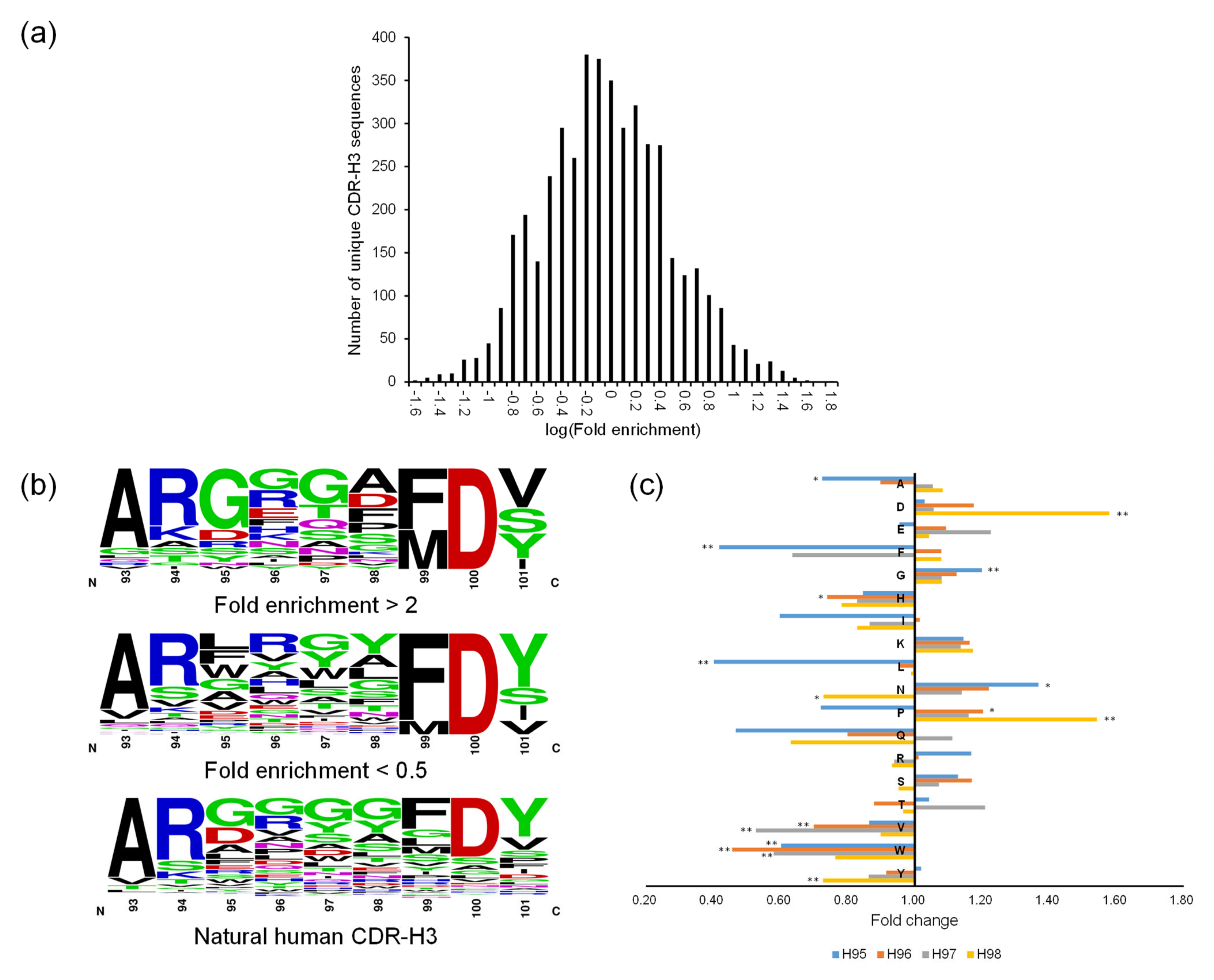
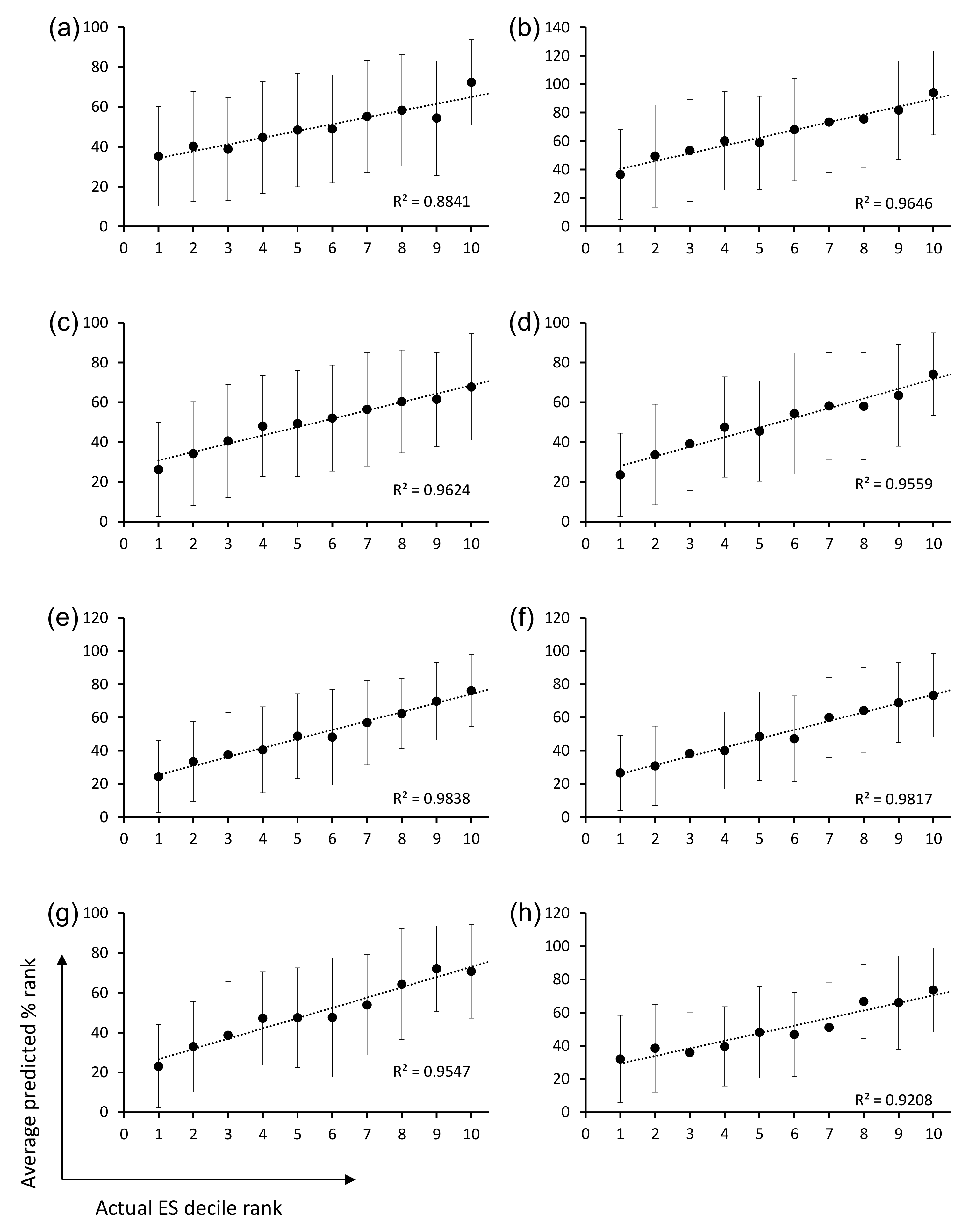
The ML models were trained and validated for CDR-H3 using the OPAL-S panning enrichment NGS data set. The trained ML models predicted the average enrichment trend of CDR-H3 sequences across the full decile range of actual ESs, although the wide variation in the prediction accuracy for individual sequences resulted in large error bars and shallow slopes (
Figure 4). The moderate accuracy of the prediction is probably due in part to the fact that the CDR-H3 sequences are unlikely to be the sole determinant of the panning amplification outcome, and also in part to the choice and the performance of ML algorithm and the quality of real-world data upon which ML models were trained and validated [
26].
In several recent studies, ML has been applied to antibody design and optimization. For example, CDR-H3 sequences with high affinity and specificity were predicted by a ML method trained on panning enrichment data against a target antigen [
27]. The high prediction accuracy of this study may be attributed in part to the fact that only CDR-H3 was diversified, thus eliminating other uncertainties within the library, and that the ML model was trained to predict a well-defined, quantifiable property (i.e., target binding affinity). In another example, ML prediction of hot spot CDR residues led to the design, construction, and validation of scFv libraries that generated antibodies to diverse epitopes on a protein antigen with affinities comparable to those of in vivo affinity-matured antibodies [
28]. The approach reported in our study utilized data obtained from a library that was panned against protein A and thus selected for amplification efficiency. While the binding specificity and affinity, which abovementioned ML-based library designs aimed to improve, depend primarily on complementary interactions between the epitope and the paratope, amplification efficiency depends on multiple factors, such as display level, protein folding, stability, and the host cell interaction of scFv clones. The complex nature of panning amplification may have contributed to the moderate prediction accuracy of the ML models. Nonetheless, the new OPAL-T library, designed and constructed based on the prediction, yielded target binders more efficiently than OPAL-S, even though the two libraries were constructed on identical frameworks and similar in size (~10
10; see above) and in the design of CDR diversity (predefined, non-combinatorial diversity). It is likely that the NGS- and ML-based optimization of the CDR sequences contributed distinctly to the improved performance of OPAL-T over OPAL-S.
The design goal of OPAL-T was to construct an antibody library with optimized amplification profile; thus, the binders were enriched faster in OPAL-T than in OPAL-S, but once individual binders were enriched by panning and isolated from the libraries, the affinities of OPAL-T clones were not significantly better than those of OPAL-S clones. For example, the average affinity (
KD) of 14 OPAL-T clones against five different antigens was 84 nM, compared with the average
KD of 29 nM for 10 OPAL-S clones against three different antigens [
3]; the average affinity of binders to SARS, for which both libraries were evaluated by SPR, was 24 nM and 34 nM for OPAL-T and OPAL-S, respectively. On the other hand, the expression and monomeric ratio of the binders isolated from OPAL-T seemed to be higher and more uniform than those from OPAL-S. Apart from different codon optimization schemes employed (see
Section 4.6) which might influence expression but not monomeric percentage, the two libraries have identical framework sequences. Since the efficient amplification of phage antibody clones requires fast growth, good expression, and/or proper folding, it is plausible that the amplification-centric design of CDR diversity also has contributed to the improved expression and aggregation resistance of OPAL-T-derived clones.
The diversity of OPAL-T was generated by massive parallel synthesis of oligonucleotides. The synthetic fidelity of nucleotide addition is very high (up to 99.5%), but for longer oligonucleotides, the full-length purity (i.e., the proportion of oligonucleotide molecules with correct length) can be much lower [
24], resulting in frameshifts and early termination of translation. In order to minimize the introduction of unintended frameshifts to the final library, single-CDR scFv libraries were prepared and subjected to a single round of panning against protein A. Although this additional selection step significantly improved the proportion of clones that were expressed solubly in the library, it also may have introduced additional bias against CDR sequences that were amplified slowly. Given the observation that the scaffold sequences used for the construction of OPAL-T appear not to have strong preference for specific CDR origins over others (except for CDR-H2 and CDR-H3, for which the diversities were designed to minimize potential biases), the biases caused by the proofread panning may be insignificant. Nonetheless, library construction will benefit by future development in oligonucleotide synthesis and purification technologies, which are expected to provide much higher synthetic fidelity.
To summarize, CDR diversities for a novel scFv library aimed at improved amplification efficiency were designed based on sequence enrichment data and ML analysis. The newly constructed OPAL-T library was validated using a panel of test antigens and demonstrated superior performance in terms of the binder enrichment efficiency compared to OPAL-S, which provided the sequencing data used for its design. Library design will be improved by more detailed analysis of the effects of various somatic hypermutations on amplification efficiency or the compatibility of diverse CDRs combined in a single scFv molecule, as well as by advances in artificial intelligence and parallel oligonucleotide synthesis technologies.
4. Materials and Methods
4.1. Materials
Oligonucleotide libraries for CDRs were synthesized by LC Sciences (Houston, TX, USA; OligomixTM). Wild-type protein A from S. aureus was purchased from Thermo Fisher Scientific (Waltham, MA, USA; PierceTM 21181). Variable domain genes for library construction were codon-optimized and synthesized (GenScript, Piscataway, NJ, US). CARS, SARS, and AIMP1 were kindly provided by Prof. Sunghoon Kim (Yonsei University, Incheon, Korea). Other reagents, antigens, and consumables used in this study were purchased from readily available commercial sources.
4.2. Panning of Phage Antibody Libraries
All experiments were performed at room temperature unless stated otherwise. An immunotube (Nunc 470319, Thermo Scientific) was coated with antigen (1–10 μg/mL in PBS) for 1 h and subsequently blocked with mPBST (PBS with 3% skim milk and 0.05% Tween-20) for another hour. Phage library (1012 cfu) preincubated for 1 h in mPBST was added to the tube and allowed to bind for 2 h. Unbound phages were removed by washing the tube with PBST (PBS with 0.05% Tween-20) three times, and bound phages were eluted by 1 mL of 100 mM triethylamine solution. The eluted phage solution was neutralized with 0.5 mL of 1 M Tris-HCl (pH 8.0), and 8.5 mL of mid-log phase TG1 E. coli culture in SB medium (Super Broth; 3% tryptone, 2% yeast extract, 1% MOPS, pH adjusted to 7.0) was added and incubated for 1 h at 37 °C with slow shaking at 120 rpm. The infected bacteria were collected by centrifugation and plated on 150-mm diameter LB-agarose plate supplemented with ampicillin (100 μg/mL) and 2% glucose (w/v).
Overnight bacterial growth was recovered from the plate by adding 5 mL of SB medium to the plate and scraping the growth with a flame-sterilized glass spreader. Fifty microliters of the collected bacterial suspension was added to 20 mL of SB medium supplemented with ampicillin (100 μg/mL). After incubating for ~2 h, when the culture became visibly turbid (OD600~0.7), 1011 pfu of VCSM13 helper phage was added. The culture was further incubated for 1 h at 37 °C with slow shaking at 120 rpm to allow helper phage superinfection. Kanamycin (70 μg/mL) was added, and the culture was incubated overnight (~16 h) at 30 °C with shaking at 220 rpm. The culture was centrifuged and the supernatant was transferred to a fresh 50 mL conical tube. Amplified phages were precipitated by adding 5 mL of 5× PEG precipitation solution (20% [w/v] PEG 8000, 15% [w/v] NaCl) and incubating the mixture on ice for >30 min. Precipitated phages were collected by centrifugation (14,000× g for 15 min), resuspended in 0.3 mL PBS, and used for the next round of selection.
4.3. NGS Analysis of OPAL-S
Sequence analysis of the pre-selection OPAL-S was described previously [
3]. For sequence analysis of the post-selection OPAL-S, phagemid DNAs from the output of the third panning round against protein A were isolated, and variable domain repertoires were amplified by PCR using a library-specific primer pair with adapter sequences for sequence analysis with Illumina MiSeq™. VH and VL sequences of the scFv library were obtained by 300 bp paired-end sequencing, and CDR sequences were extracted using an in-house Python script. Invariable framework region sequences of OPAL-S were identified in each variable domain sequence, and the sequence between two adjacent framework regions was identified as CDR. Each of the retrieved CDR sequences (except CDR-H3) was matched to the most similar human germline CDR sequence.
4.4. ML-Based Optimization of CDR-H3 Sequences
The enrichment score (ES) of each CDR-H3 sequence in the pre- and post-panning NGS repertoires was calculated using the formula:
where
npre and
npost are the read counts of the specific CDR-H3 sequence in pre- and post-panning repertoires, respectively, and
Npre and
Npost are the total read counts of the pre- and post-panning libraries, respectively.
The calculated ESs of CDR-H3 sequences were used to train ML models using a web-based ML service [
29]. Specifically, a comma-separated values (.csv) file comprising the CDR sequences, their ESs, and the amino acid residues by position was created for each CDR-H3 length to train an ML model. The CDR-H3 sequence, its individual amino acid residues, and the ES were given the attributes of text, categorical, and numeric, respectively, and the ES was set as a target. The following parameters were used: maximum ML model size, 100 MB; maximum number of data passes, 100; L2 regularization, mild (10
−6). Seventy percent of ES data was used as the training data set, and the remaining 30% was used as the evaluation data set to validate the models. Subsequently, CDR-H3 sequences simulated as described in a previous report [
3] were evaluated using the validated ML model, and sequences with predicted ES > 0 were selected for parallel oligonucleotide synthesis.
4.5. In Silico Deimmunization
The designed CDR sequences were then evaluated for their potential binding to human MHC class 2 molecules using netMHCIIpan-3.1 software [
22]. Alleles used for evaluation were DRB1*01:01, DRB1*03:01, DRB1*03:02, DRB1*04:01, DRB1*04:04, DRB1*04:05, DRB1*07:01, DRB1*08:02, DRB1*08:03, DRB1*09:01, DRB1*11:01, DRB1*13:01, DRB1*13:02, DRB1*12:02, DRB1*14:01, DRB1*15:01, DRB1*15:03, DRB3*01:01, DRB4*01:01, and DRB5*01:01, which, when combined, represent 81.2%, 75.1, 71.3, and 61.7% of Caucasian, East Asian (Korean), Black, and Hispanic populations, respectively [
23]. Overlapping 9-aa fragments of the CDR sequences (with the adjoining 8-aa framework sequences on both sides) were analyzed, and the CDR sequences predicted to be strong binders (0.5% threshold) to any of the 20 alleles were discarded.
4.6. Library Construction
The adjoining framework sequences and PCR adaptor sequences were added to both ends of the designed CDR sequences to produce a collection of 136-mer oligonucleotide sequences for parallel synthesis. In total, 27,911 sequences were synthesized (LC Sciences; purity unmeasurable due to the minute quantity and the mixed nature of the oligo pool). The oligonucleotides were amplified by PCR using a pair of primers specific for the adaptor sequences, and individual CDRs (CDR-H1-H3 and L1-L3 for kappa and lambda classes) were further amplified using primers specific for the framework sequences. For CDR-H3, PCR-amplified oligonucleotides were separated by length using PAGE.
Variable domain genes for the library were codon-optimized for human, and rare codons in
E. coli were subsequently replaced with codons non-rare in both human and
E. coli when possible. Codon-optimized VH3-23/JH4-(G4S)3-Vκ3-20/Jκ1 and VH3-23/JH4-(G4S)3-Vλ1-47/Jλ2 scaffolds were synthesized (GenScript), to which an individual CDR repertoire was inserted by overlap-extension PCR to make 16 single-CDR variant libraries (eight for CDR-H3 of different lengths, plus one each for other CDRs of heavy, kappa, and lambda variable domains). These libraries were subjected to a single round of panning against protein A to remove CDR sequences with synthetic errors that resulted in stop codons, indels, and frameshifts. The proofread CDR sequences were amplified from the panning output and assembled with framework regions by a series of overlap extension PCRs. The assembled scFv repertoires were ligated into pCOMB3X phagemid vector and transformed to electrocompetent TG1
E. coli (Lucigen, Middleton, WI, USA) to produce final phagemid libraries [
3]. PCR schemes and primer sequences for construction of the library are provided in
Tables S1–S6, respectively.
4.7. Dot Blotting
Single colonies of library clones in TG1 E. coli were grown in 200 μL of SB-ampicillin in a 96 well plate for 4 h at 37 °C. IPTG was added to each well (1 mM final concentration) and the plate was incubated overnight at 30 °C. Next day, the plate was centrifuged (2500× g for 15 min at 4 °C) and supernatants were removed. Bacterial cell pellets were resuspended in 60 μL/well of ice-cold 1× TES buffer (50 mM Tris, 1 mM EDTA, 20% [w/v] sucrose, pH 8.0). Subsequently 90 μL/well of ice-cold 0.2× TES was added and mixed well, and the plate was incubated on ice for 30 min. After centrifugation (2500× g for 15 min at 4 °C), 1 μL of the supernatant containing periplasmic extract from each well was applied to a piece of nitrocellulose membrane. The membrane was allowed to dry, blocked with mPBST, and incubated with 1:3000 dilution of anti-HA-HRP antibody (sc7392, Santa Cruz Biotechnology, Dallas, TX, USA) in mPBST for 1 h at room temperature with slow shaking. The dot-blot membrane was detected by enhanced chemiluminescence (ECL) reagent.
4.8. Purification of scFv
Binder scFv clones from panning output in TG1 E. coli were grown in 20 mL of SB-ampicillin (100 μg/mL) at 37 °C until OD600 = 0.7. IPTG was added (1 mM final concentration), and the induced culture was incubated overnight at 30 °C with shaking (200 rpm). Next day, the culture was centrifuged (2500× g for 15 min at 4 °C), supernatant was removed, and the cell pellet was resuspended in 1 mL of ice-cold 1× TES buffer. Subsequently 1.5 mL of ice-cold 0.2× TES was added and mixed well, and the mixture was incubated in ice for 30 min. After centrifugation (14,000× g for 15 min at 4 °C), the supernatant was transferred to a clean 15 mL conical tube, 5 mM final concentration of MgCl2 was added to quench EDTA, and 100 μL of Ni-NTA agarose beads suspension (EMD Millipore) was added. The mixture was slowly rotated for 1 h to allow binding of scFv to the beads at room temperature, centrifuged briefly, and the precipitated beads were washed twice with 1 mL of wash buffer (PBS with 5 mM imidazole, pH 7.4). Finally, scFv was eluted by adding 200 μL fractions of elution buffer (PBS with 200 mM imidazole, pH 7.4).
For larger scale expression and purification, 400 mL of SB-ampicillin was inoculated with 5 mL seed culture. After initial growth and overnight induction as above, periplasmic extract was obtained using 16 mL of 1× TES and 24 mL of 0.2× TES. The extract after MgCl2 treatment was applied to 0.5 mL of Ni-NTA agarose beads in a gravity flow column (Poly-Prep, Bio-Rad, Hercules, CA, USA). The beads were washed twice with 10 mL of wash buffer, and eluted in 0.5 mL fractions.
4.9. Surface Plasmon Resonance
SPR analysis was performed by using Biacore 3000 system. The antigen in sodium acetate solution (pH 4.0~5.5) was immobilized (target RU: 800~1200) on a CM5 chip (Cytiva) at a flow rate of 5 μL/min following the standard amine coupling protocol provided by the manufacturer, and purified scFv antibodies at 5–6 different concentrations were injected at a flow rate of 40 μL/min. The data was fitted to 1:1 Langmuir binding model or 1:1 binding with drifting baseline model using BiaEvaluation software to obtain kinetic parameters and dissociation constants.
4.10. Size Exclusion Chromatography
Size exclusion chromatography analysis was performed on ÄKTA Pure system (Cytiva, Marlborough, MA, USA) using Superdex 200 Increase 10/300 GL column (Cytiva). Purfied scFv (500 μL of 1–2 mg/mL concentration) was injected and run on degassed PBS buffer at a flow rate of 0.75 mL/min, and the eluted protein was monitored by absorbance at 280 nm.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}