
Dock10 Regulates Cardiac Function under Neurohormonal Stress

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
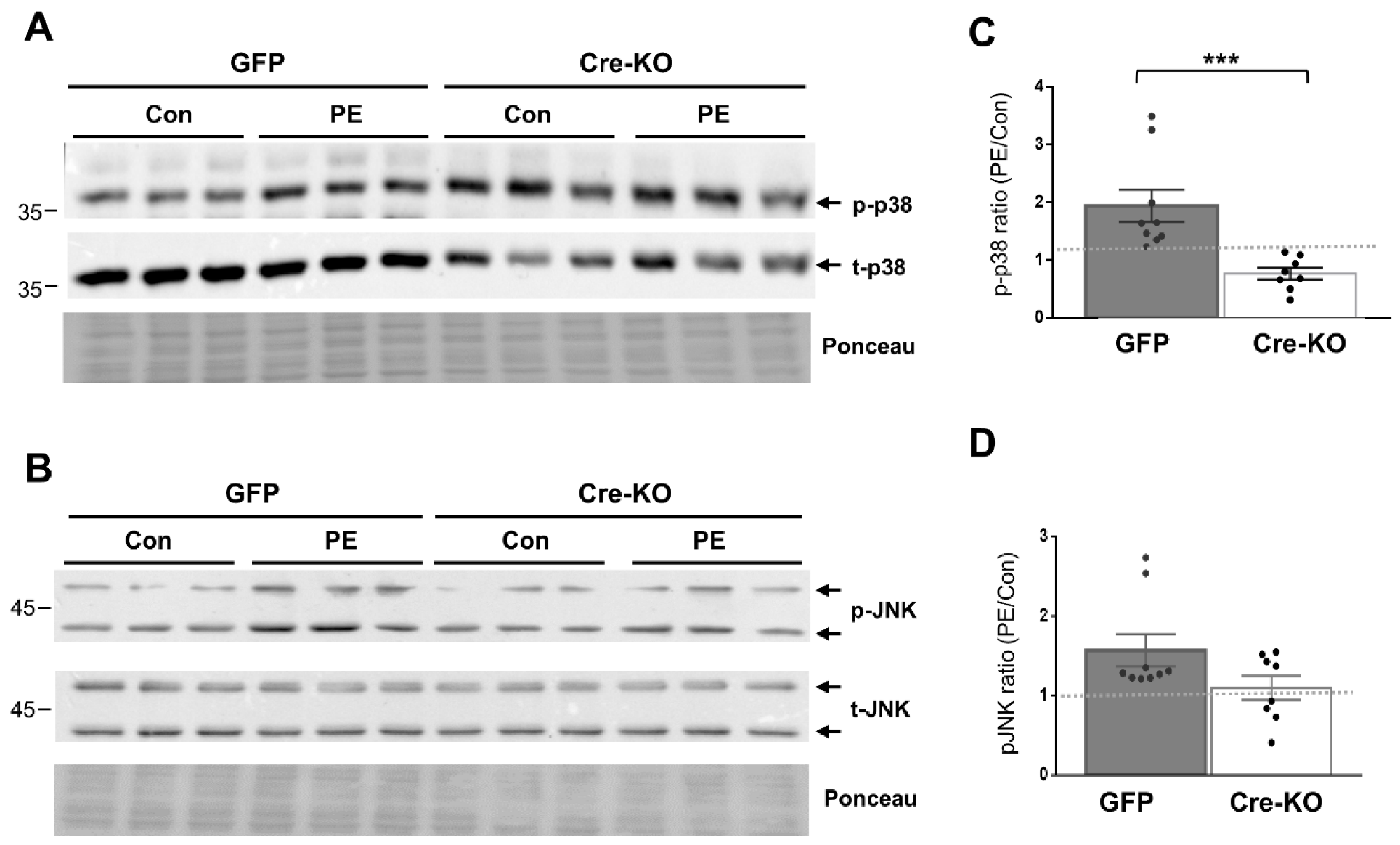
2.1. Dock10 Ablation Differentially Affects p38 and JNK Signaling in NMCMs and NMCFs
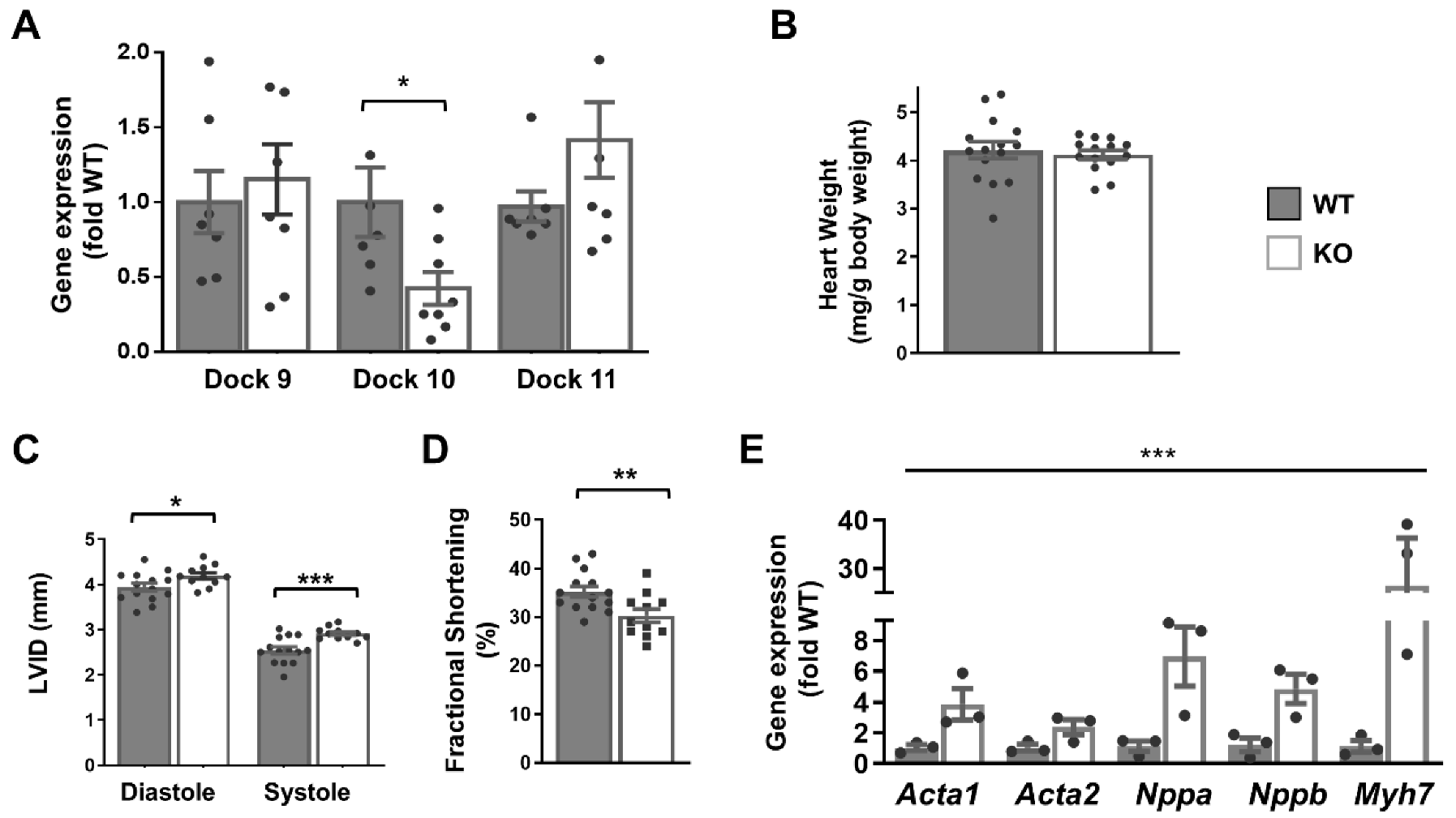
2.2. Mice with Global Dock10 KO Demonstrate a Subtle Phenotype of Cardiac Dysfunction
2.3. α-Adrenergic-Induced MAPK Signaling in the Heart Depends on Dock10 Expression
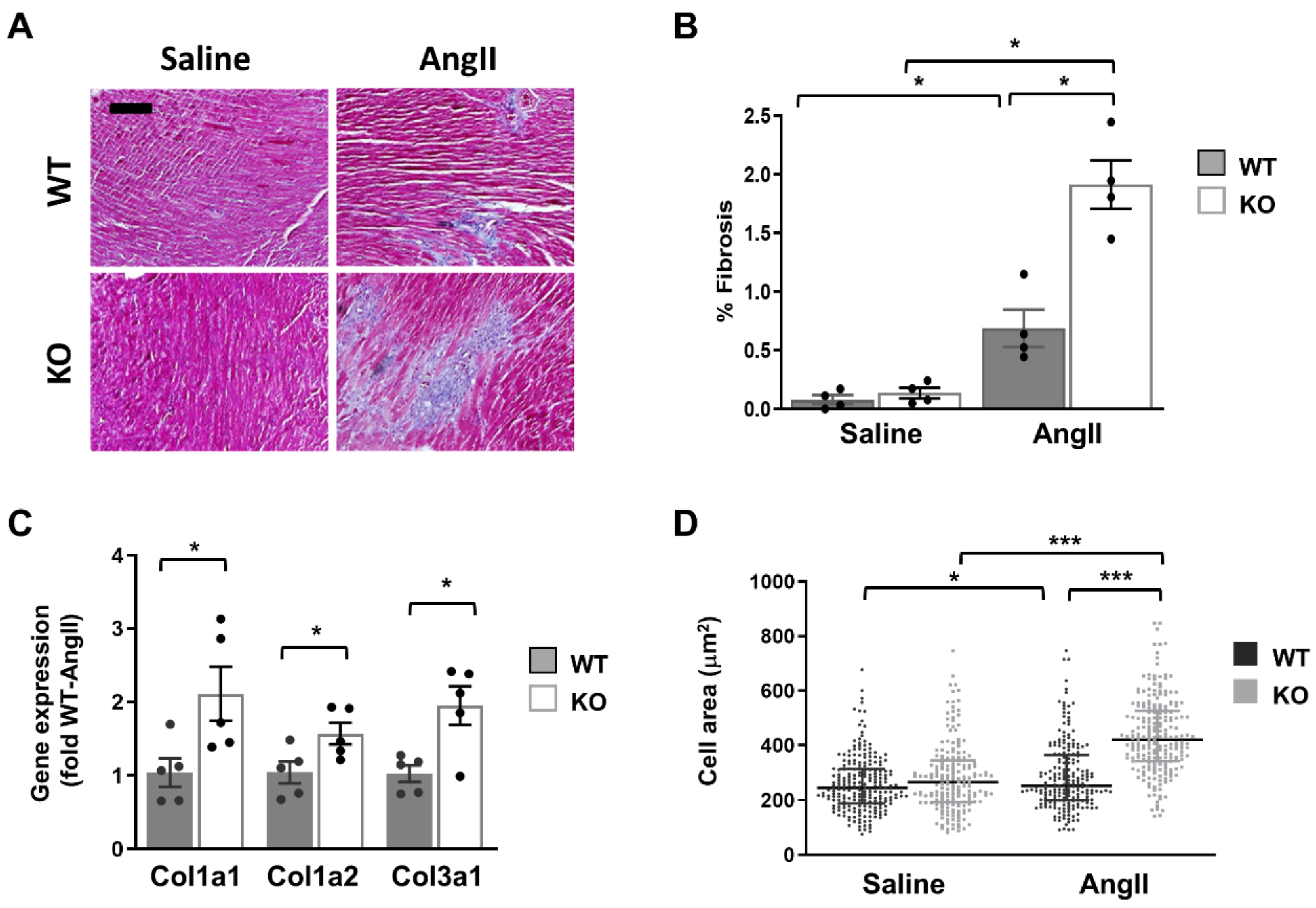
2.4. Global Dock10 KO Exacerbates Angiotensin II-Induced Pathological Cardiac Remodeling
2.5. Cardiomyocyte-Specific KO of Dock10 Does Not Exacerbate Pathological Cardiac Phenotype
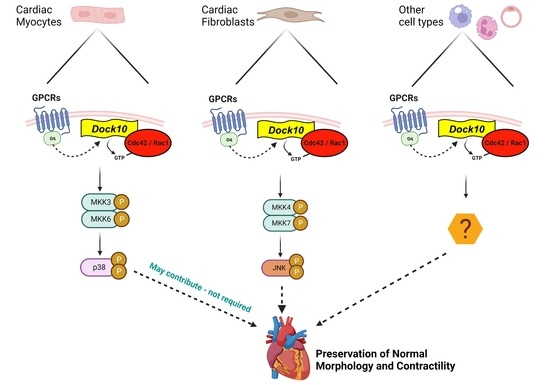
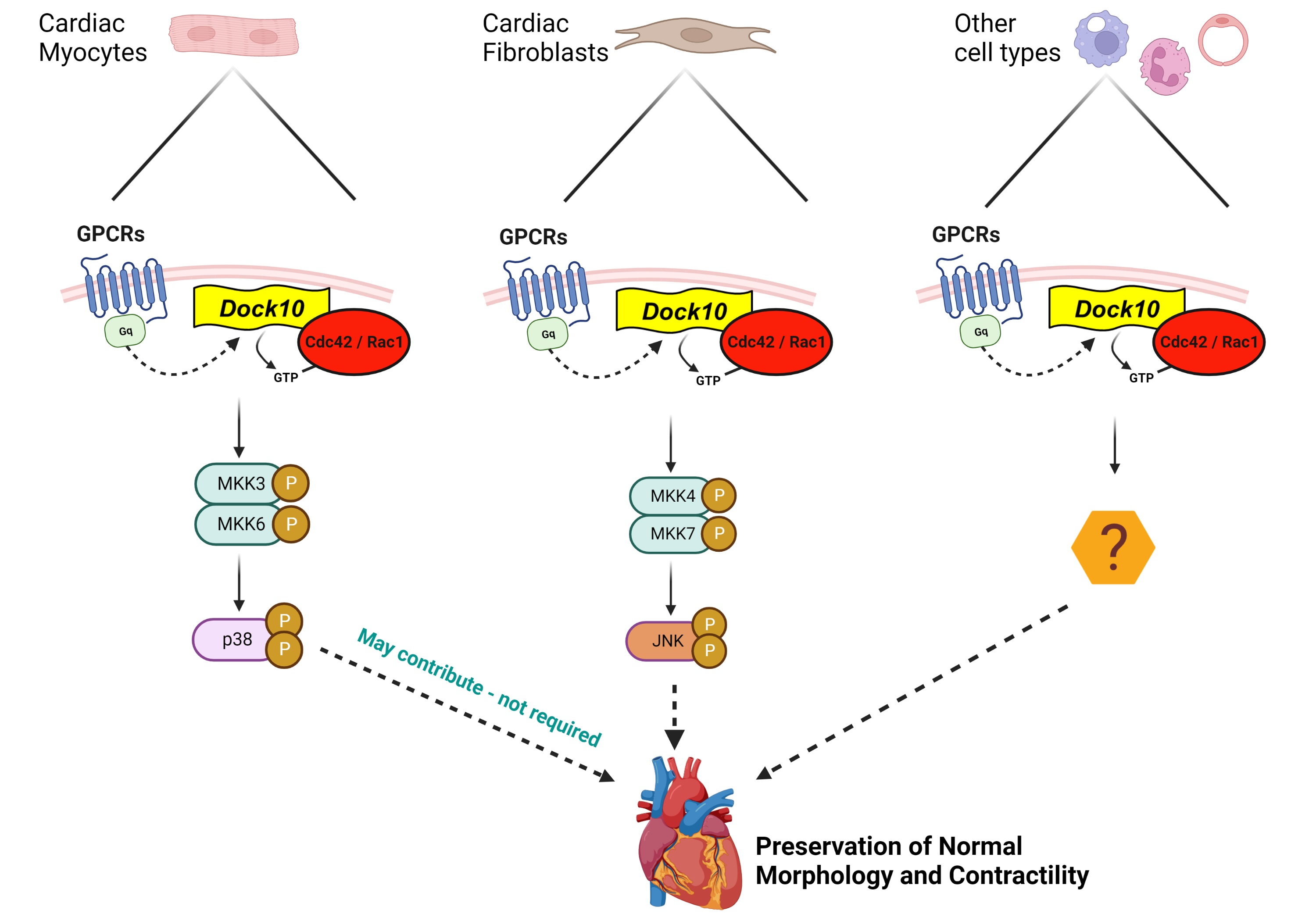
3. Discussion
4. Limitations
5. Methods
5.1. Animal Care
5.2. Global Dock10 KO Model
5.3. Dock10 CKO Model
5.4. Genotyping
5.5. Osmotic Mini Pumps
5.6. Echocardiography
5.7. Acute α-Adrenergic Stimulation
5.8. Calcium and Contractility Measurements from Isolated Cardiomyocytes
5.9. Dock10 Ablation in Primary Cultures of Neonatal Mouse-Heart Cells
5.10. Histological Analysis
5.11. Western Blotting
5.12. Gene-Expression Analysis by Quantitative PCR
5.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Berlo, J.H.; Maillet, M.; Molkentin, J.D. Signaling effectors underlying pathologic growth and remodeling of the heart. J. Clin. Investig. 2013, 123, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Burchfield, J.S.; Xie, M.; Hill, J.A. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef]
- You, J.; Wu, J.; Zhang, Q.; Ye, Y.; Wang, S.; Huang, J.; Liu, H.; Wang, X.; Zhang, W.; Bu, L.; et al. Differential cardiac hypertrophy and signaling pathways in pressure versus volume overload. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H552–H562. [Google Scholar] [CrossRef]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef]
- Lezoualc’h, F.; Metrich, M.; Hmitou, I.; Duquesnes, N.; Morel, E. Small GTP-binding proteins and their regulators in cardiac hypertrophy. J. Mol. Cell Cardiol. 2008, 44, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Lauriol, J.; Keith, K.; Jaffre, F.; Couvillon, A.; Saci, A.; Goonasekera, S.A.; McCarthy, J.R.; Kessinger, C.W.; Wang, J.; Ke, Q.; et al. RhoA signaling in cardiomyocytes protects against stress-induced heart failure but facilitates cardiac fibrosis. Sci. Signal. 2014, 7, ra100. [Google Scholar] [CrossRef]
- Ferri, N.; Contini, A.; Bernini, S.K.; Corsini, A. Role of small GTPase protein Rac1 in cardiovascular diseases: Development of new selective pharmacological inhibitors. J. Cardiovasc. Pharmacol. 2013, 62, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Ogita, H.; Takeshita, K.; Mukai, Y.; Kwiatkowski, D.J.; Liao, J.K. Requirement of Rac1 in the development of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 7432–7437. [Google Scholar] [CrossRef]
- Maillet, M.; Lynch, J.M.; Sanna, B.; York, A.J.; Zheng, Y.; Molkentin, J.D. Cdc42 is an antihypertrophic molecular switch in the mouse heart. J. Clin. Investig. 2009, 119, 3079–3088. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Bueno, O.F.; Wilkins, B.J.; Kuan, C.Y.; Xia, Y.; Molkentin, J.D. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J. 2003, 22, 5079–5089. [Google Scholar] [CrossRef]
- Chow, C.W.; Rincon, M.; Cavanagh, J.; Dickens, M.; Davis, R.J. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science 1997, 278, 1638–1641. [Google Scholar] [CrossRef]
- Ruiz-Lafuente, N.; Alcaraz-Garcia, M.J.; Garcia-Serna, A.M.; Sebastian-Ruiz, S.; Moya-Quiles, M.R.; Garcia-Alonso, A.M.; Parrado, A. Dock10, a Cdc42 and Rac1 GEF, induces loss of elongation, filopodia, and ruffles in cervical cancer epithelial HeLa cells. Biol. Open 2015, 4, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Jaudon, F.; Raynaud, F.; Wehrle, R.; Bellanger, J.M.; Doulazmi, M.; Vodjdani, G.; Gasman, S.; Fagni, L.; Dusart, I.; Debant, A.; et al. The RhoGEF DOCK10 is essential for dendritic spine morphogenesis. Mol. Biol. Cell 2015, 26, 2112–2127. [Google Scholar] [CrossRef]
- Yelo, E.; Bernardo, M.V.; Gimeno, L.; Alcaraz-Garcia, M.J.; Majado, M.J.; Parrado, A. Dock10, a novel CZH protein selectively induced by interleukin-4 in human B lymphocytes. Mol. Immunol. 2008, 45, 3411–3418. [Google Scholar] [CrossRef]
- Alcaraz-García, M.J.; Ruiz-Lafuente, N.; Sebastián-Ruiz, S.; Majado, M.J.; González-García, C.; Bernardo, M.V.; Alvarez-López, M.R.; Parrado, A. Human and mouse DOCK10 splicing isoforms with alternative first coding exon usage are differentially expressed in T and B lymphocytes. Hum. Immunol. 2011, 72, 531–537. [Google Scholar] [CrossRef]
- García-Serna, A.M.; Alcaraz-García, M.J.; Ruiz-Lafuente, N.; Sebastián-Ruiz, S.; Martínez, C.M.; Moya-Quiles, M.R.; Minguela, A.; García-Alonso, A.M.; Martín-Orozco, E.; Parrado, A. Dock10 regulates CD23 expression and sustains B-cell lymphopoiesis in secondary lymphoid tissue. Immunobiology 2016, 221, 1343–1350. [Google Scholar] [CrossRef]
- Matsuda, T.; Yanase, S.; Takaoka, A.; Maruyama, M. The immunosenescence-related gene Zizimin2 is associated with early bone marrow B cell development and marginal zone B cell formation. Immun. Ageing 2015, 12, 1. [Google Scholar] [CrossRef]
- Gerasimčik, N.; He, M.; Baptista, M.A.P.; Severinson, E.; Westerberg, L.S. Deletion of Dock10 in B Cells Results in Normal Development but a Mild Deficiency upon In Vivo and In Vitro Stimulations. Front. Immunol. 2017, 8, 491. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Matsuda, T.; Kawaguchi, K.; Takaoka, A.; Maruyama, M. Involvement of Zizimin2/3 in the age-related defect of peritoneal B-1a cells as a source of anti-bacterial IgM. Int. Immunol. 2017, 29, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; Sanz-Moreno, V.; Self, A.; Godi, A.; Marshall, C.J. DOCK10-mediated Cdc42 activation is necessary for amoeboid invasion of melanoma cells. Curr. Biol. 2008, 18, 1456–1465. [Google Scholar] [CrossRef]
- Kritzer, M.D.; Li, J.; Passariello, C.L.; Gayanilo, M.; Thakur, H.; Dayan, J.; Dodge-Kafka, K.; Kapiloff, M.S. The scaffold protein muscle A-kinase anchoring protein beta orchestrates cardiac myocyte hypertrophic signaling required for the development of heart failure. Circ. Heart Fail. 2014, 7, 663–672. [Google Scholar] [CrossRef]
- Hall, A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005, 33 Pt. 5, 891–895. [Google Scholar] [CrossRef]
- Minden, A.; Lin, A.; Claret, F.X.; Abo, A.; Karin, M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 1995, 81, 1147–1157. [Google Scholar] [CrossRef]
- Coso, O.A.; Chiariello, M.; Yu, J.C.; Teramoto, H.; Crespo, P.; Xu, N.; Miki, T.; Gutkind, J.S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 1995, 81, 1137–1146. [Google Scholar] [CrossRef]
- Shin, S.; Buel, G.R.; Nagiec, M.J.; Han, M.J.; Roux, P.P.; Blenis, J.; Yoon, S.O. ERK2 regulates epithelial-to-mesenchymal plasticity through DOCK10-dependent Rac1/FoxO1 activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2967–2976. [Google Scholar] [CrossRef]
- Ruiz-Lafuente, N.; Minguela, A.; Muro, M.; Parrado, A. The role of DOCK10 in the regulation of the transcriptome and aging. Heliyon 2019, 5, e01391. [Google Scholar] [CrossRef] [PubMed]
- Namekata, K.; Guo, X.; Kimura, A.; Azuchi, Y.; Kitamura, Y.; Harada, C.; Harada, T. Roles of the DOCK-D family proteins in a mouse model of neuroinflammation. J. Biol. Chem. 2020, 295, 6710–6720. [Google Scholar] [CrossRef]
- Zhang, S.; Han, J.; Sells, M.A.; Chernoff, J.; Knaus, U.G.; Ulevitch, R.J.; Bokoch, G.M. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J. Biol. Chem. 1995, 270, 23934–23936. [Google Scholar] [CrossRef]
- Philips, A.; Roux, P.; Coulon, V.; Bellanger, J.M.; Vié, A.; Vignais, M.L.; Blanchard, J.M. Differential effect of Rac and Cdc42 on p38 kinase activity and cell cycle progression of nonadherent primary mouse fibroblasts. J. Biol. Chem. 2000, 275, 5911–5917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samani, A.; English, K.G.; Lopez, M.A.; Birch, C.L.; Brown, D.M.; Kaur, G.; Worthey, E.A.; Alexander, M.S. DOCKopathies: A systematic review of the clinical pathologies associated with human DOCK pathogenic variants. Hum. Mutat. 2022, 43, 1149–1161. [Google Scholar] [CrossRef]
- Benson, C.E.; Southgate, L. The DOCK protein family in vascular development and disease. Angiogenesis 2021, 24, 417–433. [Google Scholar] [CrossRef]
- Taniike, M.; Yamaguchi, O.; Tsujimoto, I.; Hikoso, S.; Takeda, T.; Nakai, A.; Omiya, S.; Mizote, I.; Nakano, Y.; Higuchi, Y.; et al. Apoptosis signal-regulating kinase 1/p38 signaling pathway negatively regulates physiological hypertrophy. Circulation 2008, 117, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Altered cardiac myocyte Ca regulation in heart failure. Physiology 2006, 21, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Kho, C.; Lee, A.; Hajjar, R.J. Altered sarcoplasmic reticulum calcium cycling—Targets for heart failure therapy. Nat. Rev. Cardiol. 2012, 9, 717–733. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Tian, X.; Liu, Y.; Song, H.; Cheng, X.; Zhang, X.; Yan, C.A.-O.; Han, Y.A.-O. CREG ameliorates the phenotypic switching of cardiac fibroblasts after myocardial infarction via modulation of CDC42. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Lavall, D.; Schuster, P.; Jacobs, N.; Kazakov, A.; Böhm, M.; Laufs, U. Rac1 GTPase regulates 11β hydroxysteroid dehydrogenase type 2 and fibrotic remodeling. J. Biol. Chem. 2017, 292, 7542–7553. [Google Scholar] [CrossRef]
- Sundararaman, A.A.-O.; Mellor, H. A functional antagonism between RhoJ and Cdc42 regulates fibronectin remodelling during angiogenesis. Small GTPases 2021, 12, 241–245. [Google Scholar] [CrossRef]
- Kempers, L.A.-O.; Driessen, A.J.M.; van Rijssel, J.; Nolte, M.A.-O.; van Buul, J.A.-O. The RhoGEF Trio: A Protein with a Wide Range of Functions in the Vascular Endothelium. Int. J. Mol. Sci. 2021, 22, 10168. [Google Scholar] [CrossRef]
- Frieler, R.A.; Mortensen, R.M. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 2015, 131, 1019–1030. [Google Scholar] [CrossRef] [Green Version]
- Aoki, H.; Richmond, M.; Izumo, S.; Sadoshima, J. Specific role of the extracellular signal-regulated kinase pathway in angiotensin II-induced cardiac hypertrophy in vitro. Biochem. J. 2000, 347 Pt 1, 275–284. [Google Scholar] [CrossRef]
- Lazou, A.; Sugden, P.H.; Clerk, A. Activation of mitogen-activated protein kinases (p38-MAPKs, SAPKs/JNKs and ERKs) by the G-protein-coupled receptor agonist phenylephrine in the perfused rat heart. Biochem. J. 1998, 332 Pt 2, 459–465. [Google Scholar] [CrossRef]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T.; et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Bejerano, T.; Etzion, S.; Elyagon, S.; Etzion, Y.; Cohen, S. Nanoparticle Delivery of miRNA-21 Mimic to Cardiac Macrophages Improves Myocardial Remodeling after Myocardial Infarction. Nano Lett. 2018, 18, 5885–5891. [Google Scholar] [CrossRef] [PubMed]
- Etzion, S.; Etzion, Y.; DeBosch, B.; Crawford, P.A.; Muslin, A.J. Akt2 deficiency promotes cardiac induction of Rab4a and myocardial beta-adrenergic hypersensitivity. J. Mol. Cell Cardiol. 2010, 49, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segal, L.; Etzion, S.; Elyagon, S.; Shahar, M.; Klapper-Goldstein, H.; Levitas, A.; Kapiloff, M.S.; Parvari, R.; Etzion, Y. Dock10 Regulates Cardiac Function under Neurohormonal Stress. Int. J. Mol. Sci. 2022, 23, 9616. https://doi.org/10.3390/ijms23179616
Segal L, Etzion S, Elyagon S, Shahar M, Klapper-Goldstein H, Levitas A, Kapiloff MS, Parvari R, Etzion Y. Dock10 Regulates Cardiac Function under Neurohormonal Stress. International Journal of Molecular Sciences. 2022; 23(17):9616. https://doi.org/10.3390/ijms23179616
Chicago/Turabian StyleSegal, Liad, Sharon Etzion, Sigal Elyagon, Moran Shahar, Hadar Klapper-Goldstein, Aviva Levitas, Michael S. Kapiloff, Ruti Parvari, and Yoram Etzion. 2022. "Dock10 Regulates Cardiac Function under Neurohormonal Stress" International Journal of Molecular Sciences 23, no. 17: 9616. https://doi.org/10.3390/ijms23179616
APA StyleSegal, L., Etzion, S., Elyagon, S., Shahar, M., Klapper-Goldstein, H., Levitas, A., Kapiloff, M. S., Parvari, R., & Etzion, Y. (2022). Dock10 Regulates Cardiac Function under Neurohormonal Stress. International Journal of Molecular Sciences, 23(17), 9616. https://doi.org/10.3390/ijms23179616