
Natural Xylooligosaccharides Exert Antitumor Activity via Modulation of Cellular Antioxidant State and TLR4

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
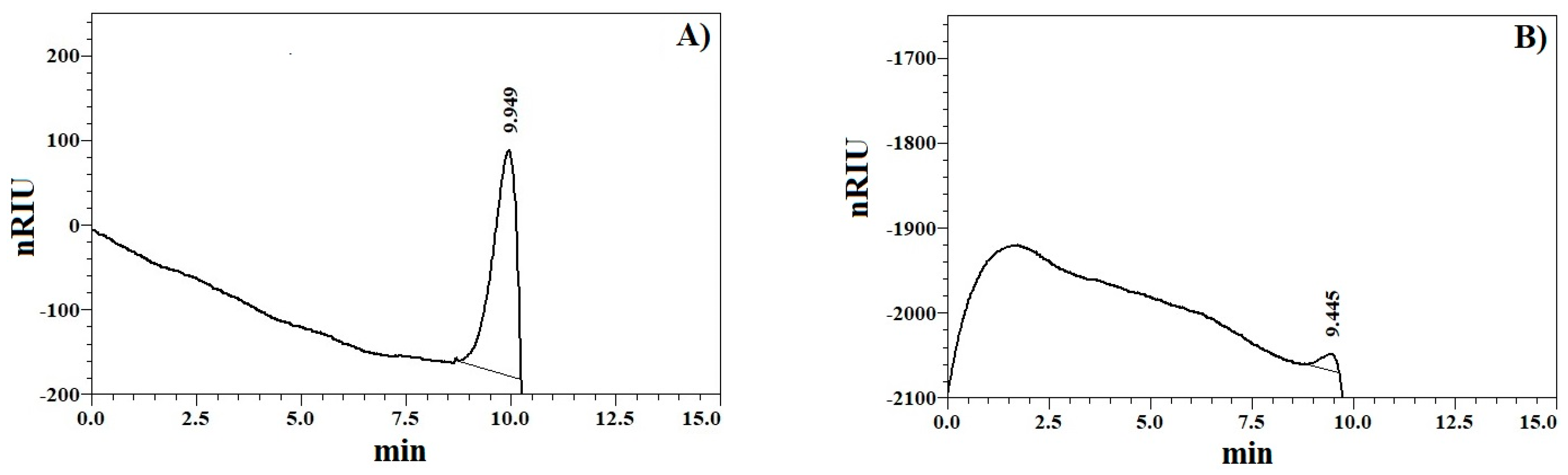
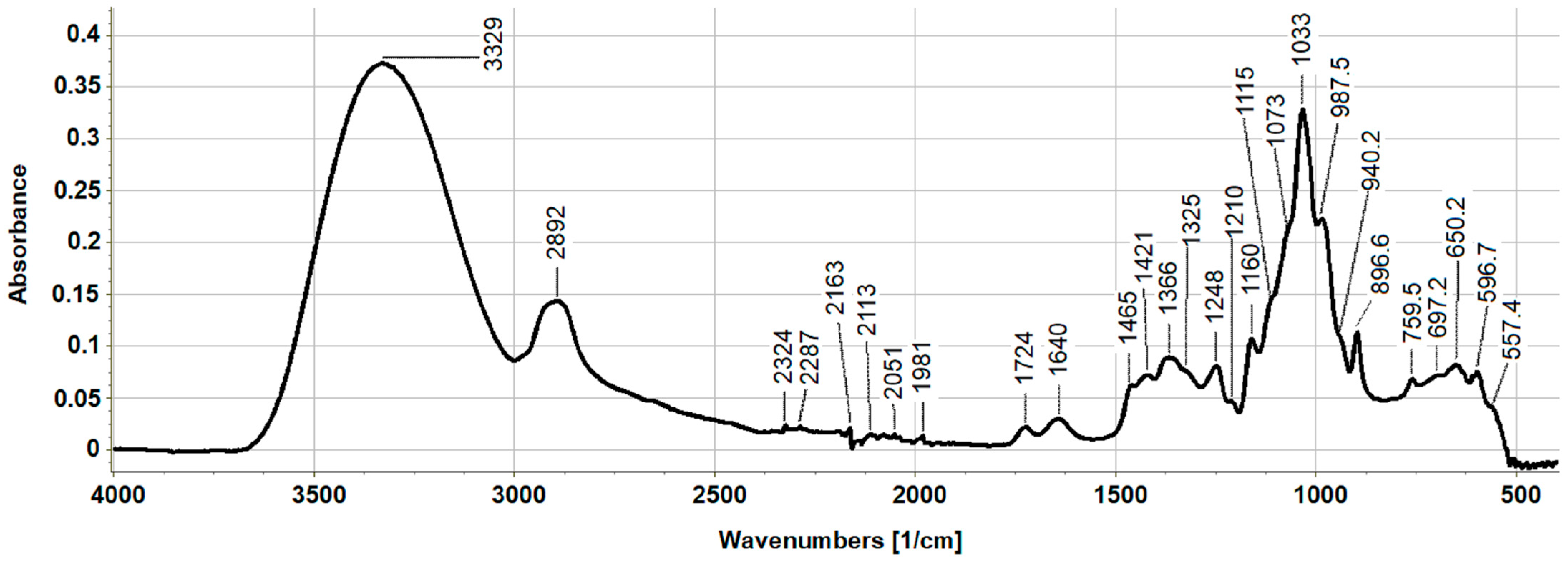
2.1. Chemical Characterization of the XOS Sample
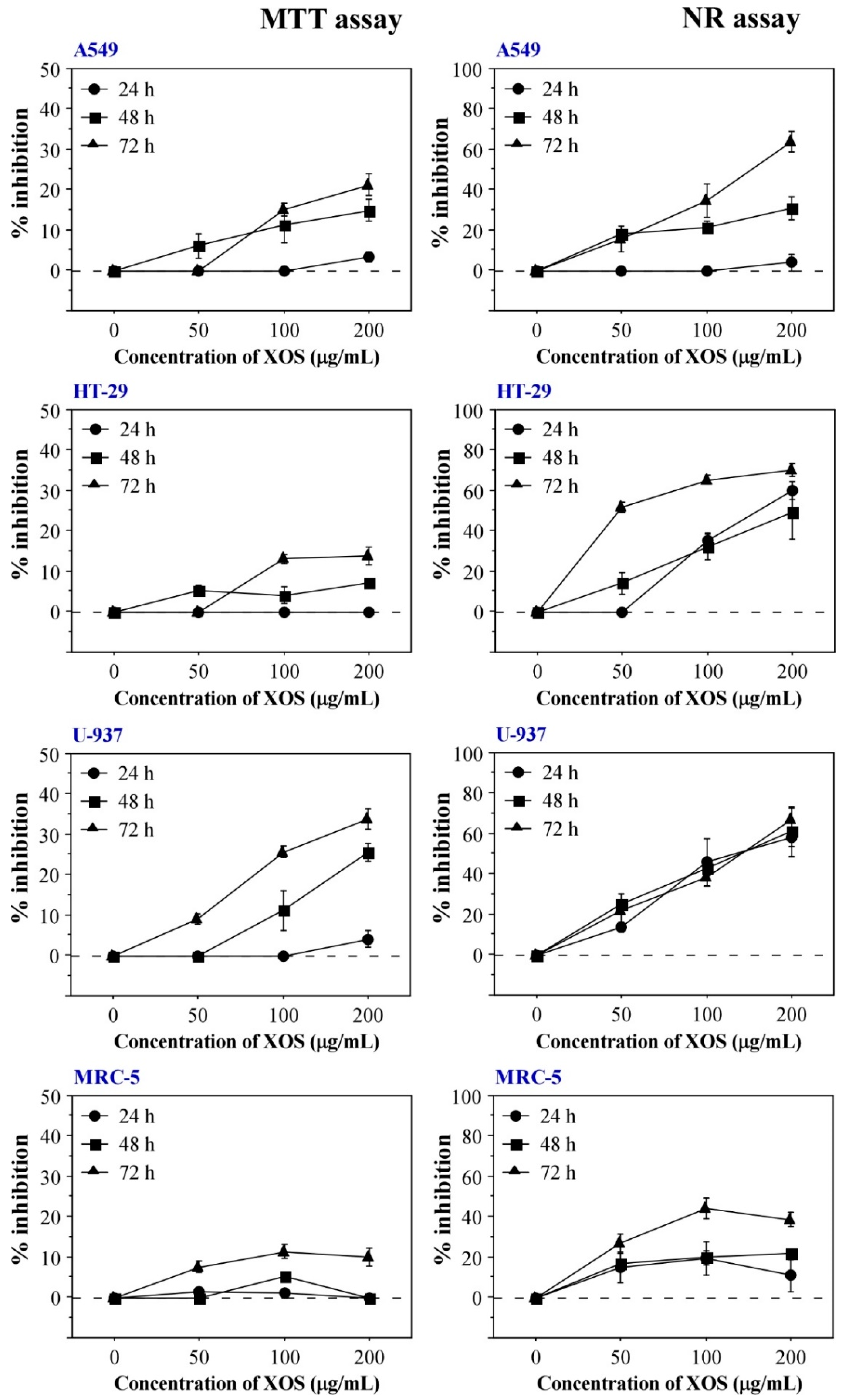
2.2. XOS Cytotoxicity and Antitumor Activity In Vitro
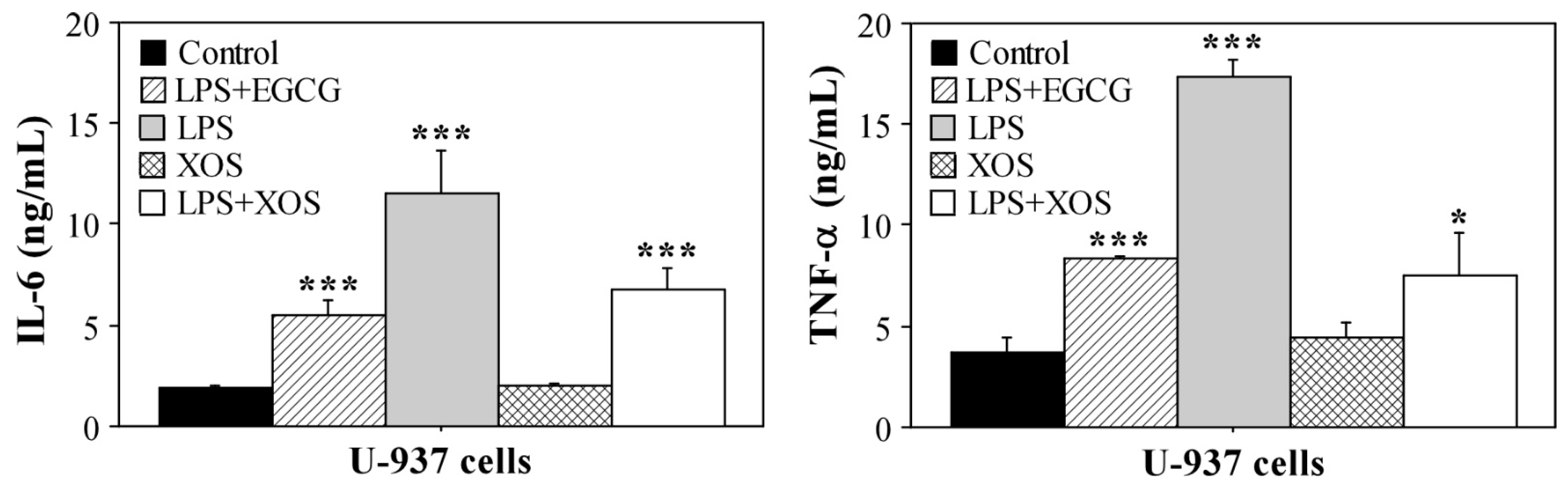
2.3. XOS Reduce Proinflammatory Cytokines Production by U-937 Cells
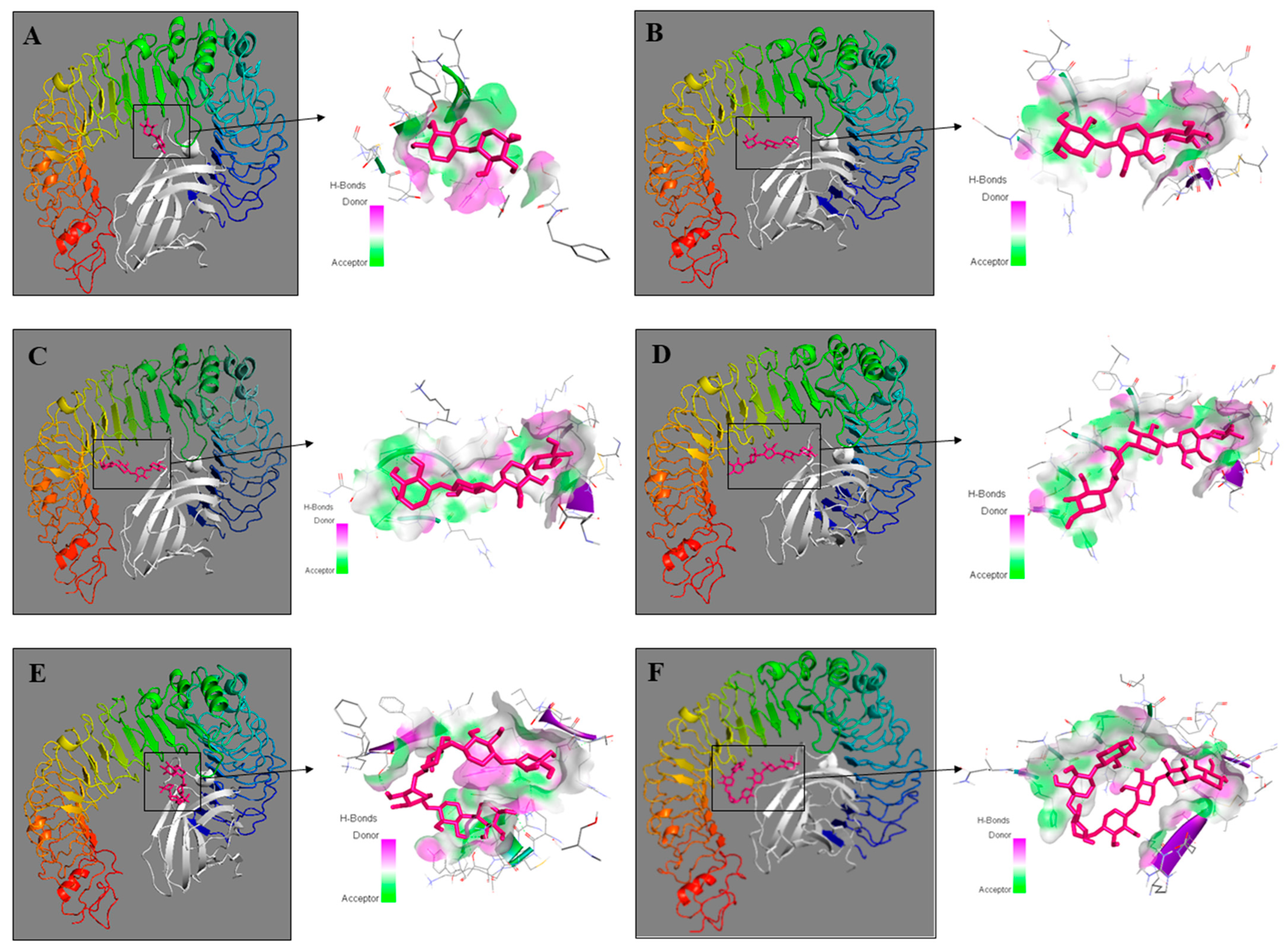
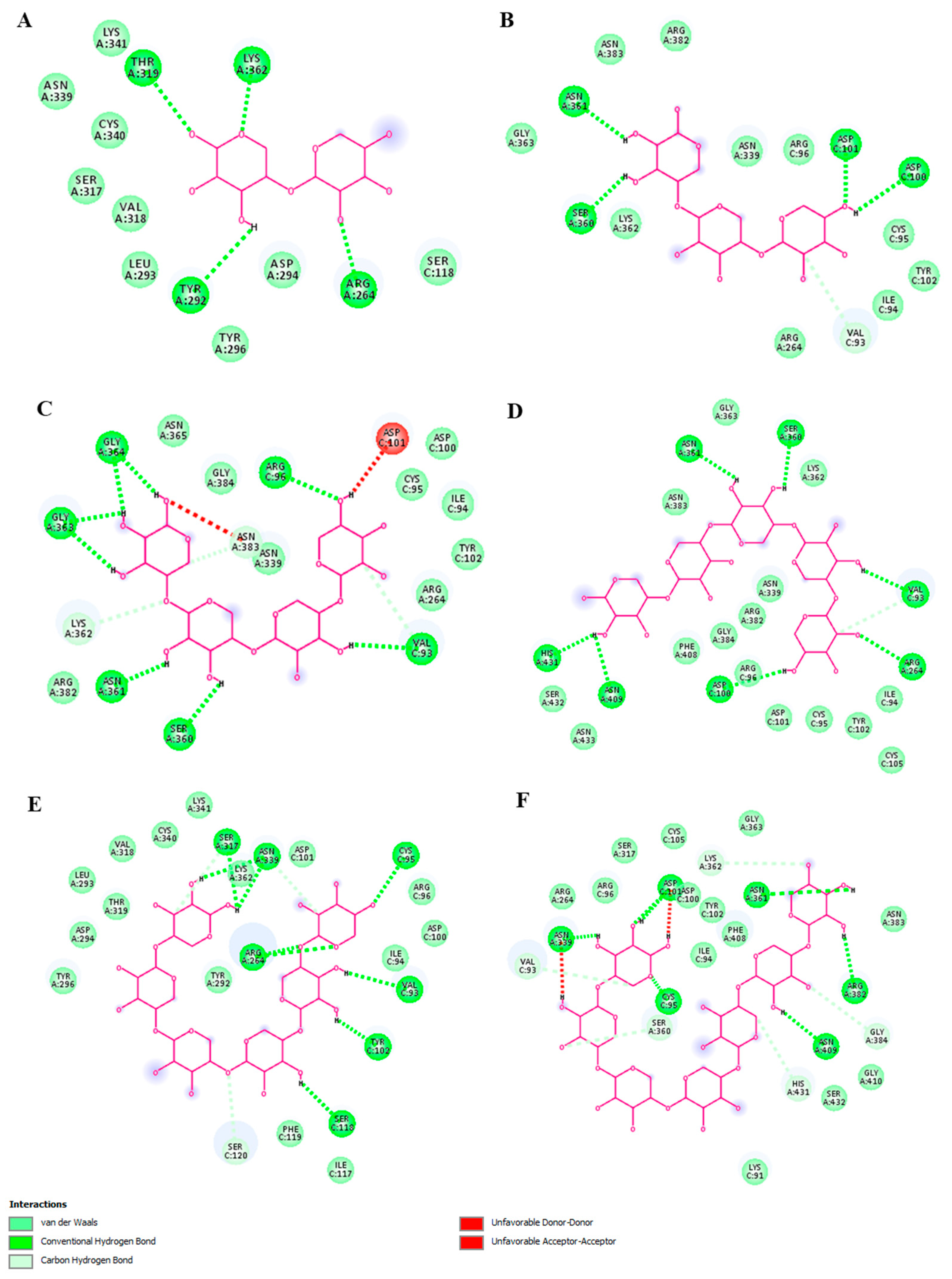
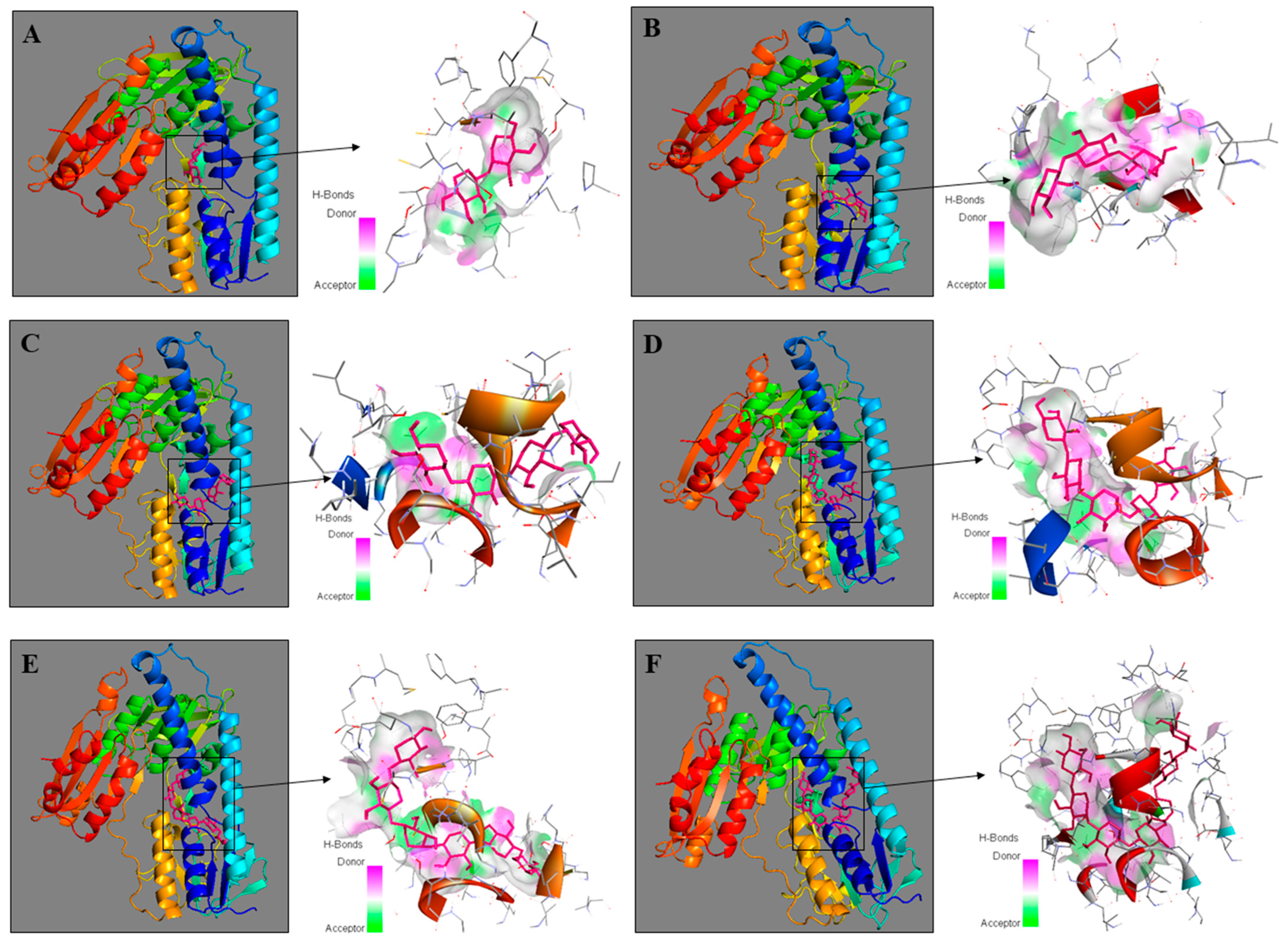
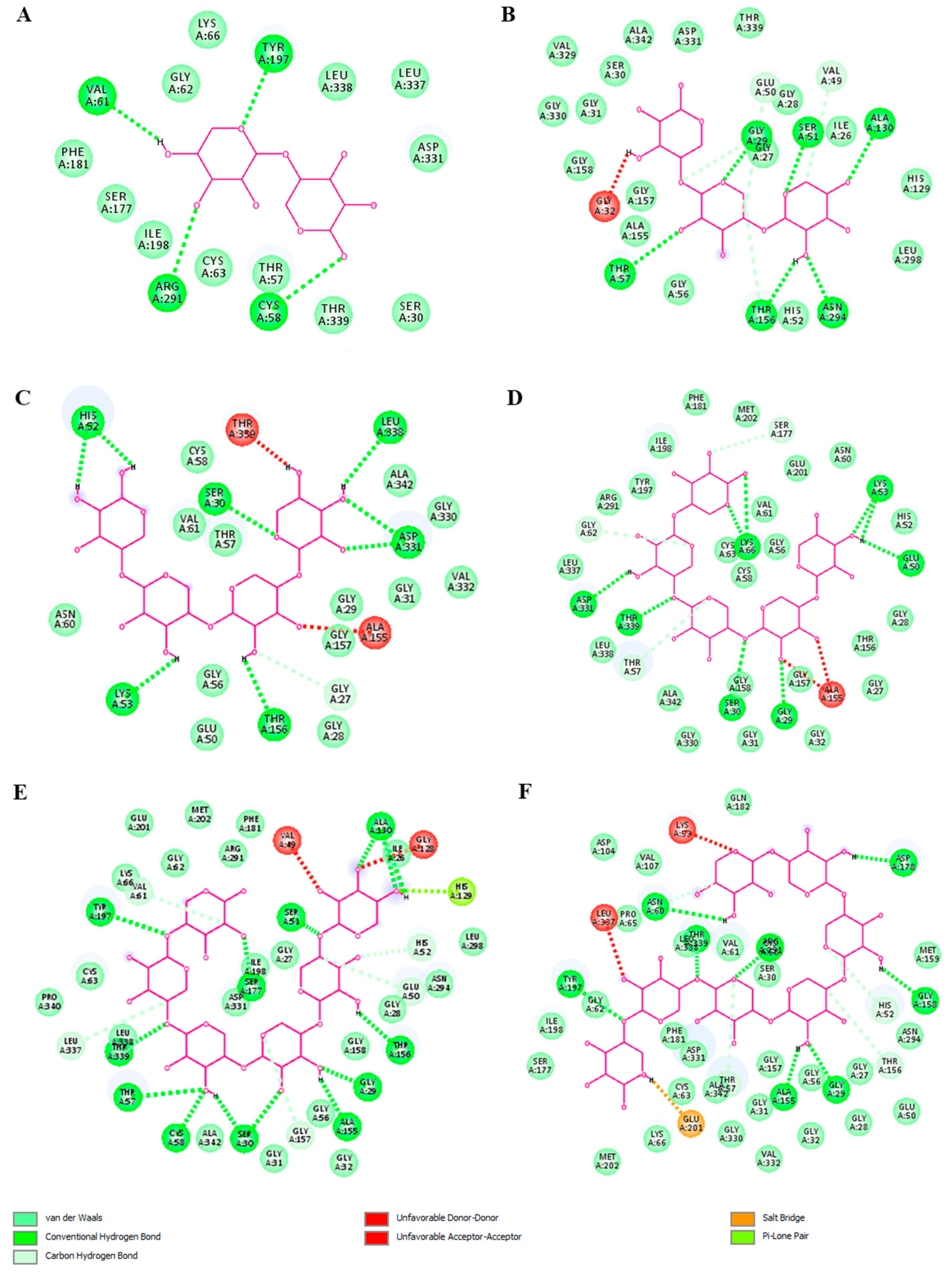
2.4. Molecular Modeling of XOS-TLR4 Interaction
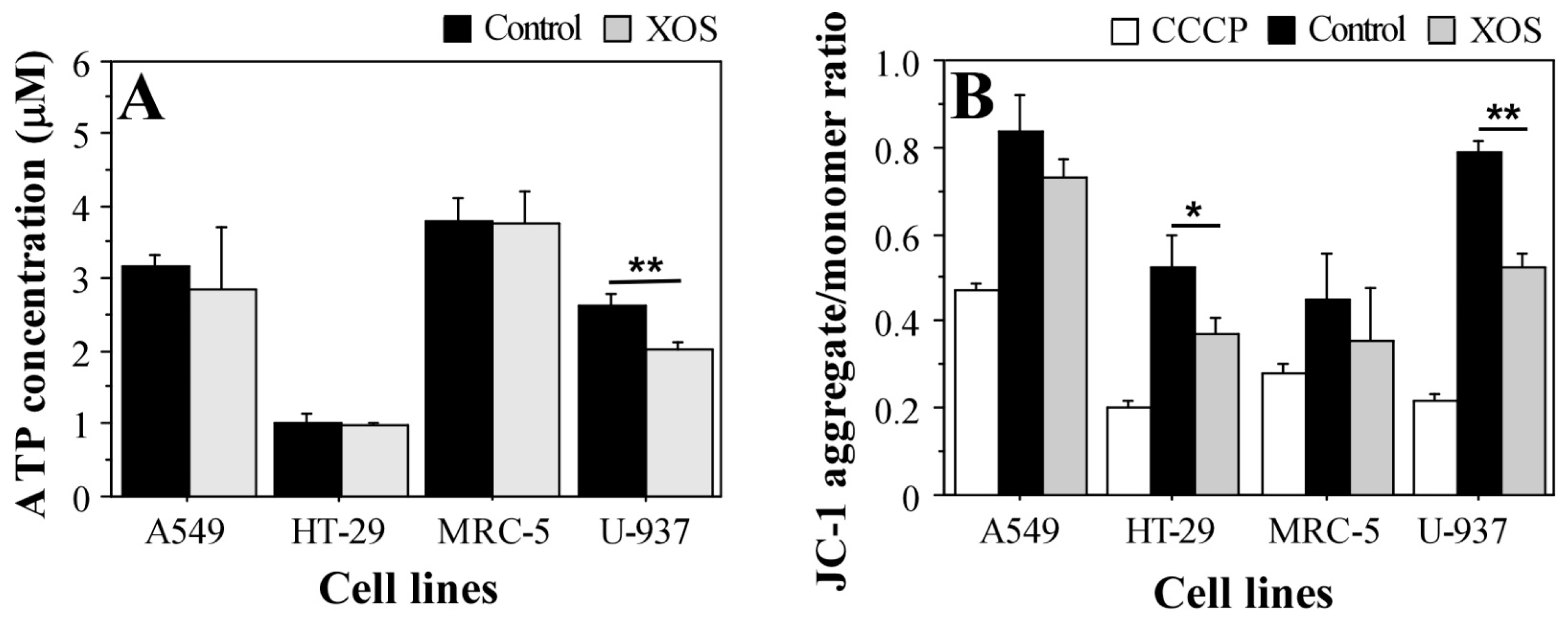
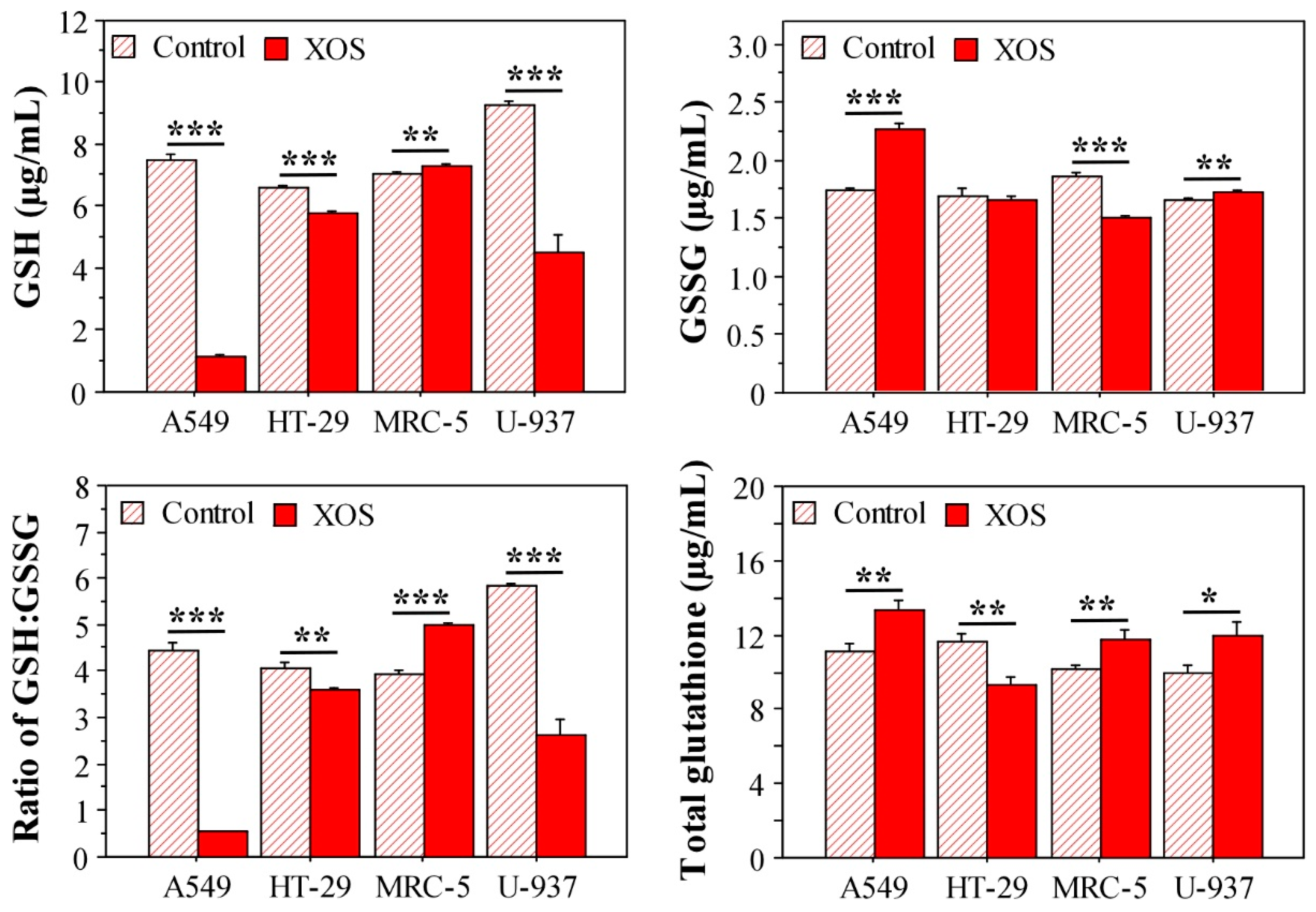
2.5. Xylooligosaccharide Treatment Affects Glutathione Homeostasis in Tumor Cells
3. Discussion
4. Materials and Methods
4.1. Xylooligosaccharides
4.2. General Chemical Analyses
4.3. Monosaccharide Composition Analysis
4.4. Molecular Weight Distribution Analysis
4.5. Fourier Transform Infrared Spectroscopy
4.6. In Vitro Antioxidant Activity
4.7. Cell Lines and In Vitro Culture Conditions
4.8. In Vitro Cytotoxicity and Antitumor Activity Assays
4.9. Evaluation of Cytokine Levels
4.10. ATP Determination
4.11. JC-1 Staining
4.12. Molecular Docking
4.13. Assessment of Glutathione Levels
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moure, A.; Gullón, P.; Domínguez, H.; Parajó, J.C. Advances in the manufacture, purification and applications of xylo-oligosaccharides as food additives and nutraceuticals. Process Biochem. 2006, 41, 1913–1923. [Google Scholar] [CrossRef]
- Wierzbicki, M.P.; Maloney, V.; Mizrachi, E.; Myburg, A.A. Xylan in the Middle: Understanding Xylan Biosynthesis and Its Metabolic Dependencies Toward Improving Wood Fiber for Industrial Processing. Front. Plant Sci. 2019, 10, 176. [Google Scholar] [CrossRef] [PubMed]
- De Freitas, C.; Carmona, E.; Brienzo, M. Xylooligosaccharides production process from lignocellulosic biomass and bioactive effects. Bioact. Carbohydr. Diet. Fibre 2019, 18, 100184. [Google Scholar] [CrossRef]
- Ebringerová, A.; Hromádková, Z.; Heinze, T. Hemicellulose. In Polysaccharides I. Structure, Characterization and Use; Heinze, T., Ed.; Springer-Verlag GmBH: Berlin/Heidelberg, Germany, 2005; pp. 1–67. [Google Scholar]
- Santibanez, L.; Henriquez, C.; Corro-Tejeda, R.; Bernal, S.; Armijo, B.; Salazar, O. Xylooligosaccharides from lignocellulosic biomass: A comprehensive review. Carbohydr. Polym. 2021, 251, 117118. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Biely, P.; Uhliarikova, I.; Sato, N.; Makishima, S.; Mizuno, M.; Nozaki, K.; Kaneko, S.; Amano, Y. Structural characterization of hemicellulose released from corn cob in continuous flow type hydrothermal reactor. J. Biosci. Bioeng. 2019, 127, 222–230. [Google Scholar] [CrossRef]
- Isci, A.; Thieme, N.; Lamp, A.; Zverlov, V.; Kaltschmitt, M. Production of xylo-oligosaccharides from wheat straw using microwave assisted deep eutectic solvent pretreatment. Ind. Crops Prod. 2021, 164, 113393. [Google Scholar] [CrossRef]
- Pattarapisitporn, A.; Thiangthong, N.; Inthajak, P.; Jaichakan, P.; Panpa, W.; Klangpetch, W. Production of Xyloligosaccharides from Rice Straw by Microwave-assisted Enzymatic Hydrolysis and Evaluation of Their Prebiotic Properties. Chiang Mai Univ. J. Nat. Sci. 2021, 20, e2021037. [Google Scholar] [CrossRef]
- Samanta, A.K.; Jayapal, N.; Jayaram, C.; Roy, S.; Kolte, A.P.; Senani, S.; Sridhar, M. Xylooligosaccharides as prebiotics from agricultural by-products: Production and applications. Bioact. Carbohydr. Diet. Fibre 2015, 5, 62–71. [Google Scholar] [CrossRef]
- Bian, J.; Peng, F.; Peng, X.P.; Peng, P.; Xu, F.; Sun, R.C. Structural features and antioxidant activity of xylooligosaccharides enzymatically produced from sugarcane bagasse. Bioresour. Technol. 2013, 127, 236–241. [Google Scholar] [CrossRef]
- Amorim, C.; Silverio, S.C.; Prather, K.L.J.; Rodrigues, L.R. From lignocellulosic residues to market: Production and commercial potential of xylooligosaccharides. Biotechnol. Adv. 2019, 37, 107397. [Google Scholar] [CrossRef] [Green Version]
- Rashid, R.; Sohail, M. Xylanolytic Bacillus species for xylooligosaccharides production: A critical review. Bioresour. Bioprocess. 2021, 8, 16. [Google Scholar] [CrossRef]
- Shimoda, K.; Hamada, H.; Hamada, H. Synthesis of xylooligosaccharides of daidzein and their anti-oxidant and anti-allergic activities. Int. J. Mol. Sci. 2011, 12, 5616. [Google Scholar] [CrossRef] [PubMed]
- Boonchuay, P.; Wongpoomchai, R.; Jaturasitha, S.; Mahatheeranont, S.; Watanabe, M.; Chaiyaso, T. Prebiotic properties, antioxidant activity, and acute oral toxicity of xylooligosaccharides derived enzymatically from corncob. Food Biosci. 2021, 40, 100895. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, Y.; Ajuwon, K.M.; Zhong, R.; Li, T.; Chen, L.; Zhang, H.; Beckers, Y.; Everaert, N. Xylo-Oligosaccharides, Preparation and Application to Human and Animal Health: A Review. Front. Nutr. 2021, 8, 731930. [Google Scholar] [CrossRef]
- Cardoso, B.B.; Amorim, C.; Silvério, S.C.; Rodrigues, L.R. Novel and emerging prebiotics: Advances and opportunities. In Advances in Food and Nutrition Research; Academic Press: Cambridge, MA, USA, 2021; pp. 41–95. [Google Scholar]
- Iliev, I.; Vasileva, T.; Bivolarski, V.; Momchilova, A.; Ivanova, I. Metabolic Profiling of Xylooligosaccharides by Lactobacilli. Polymers 2020, 12, 2387. [Google Scholar] [CrossRef]
- Finegold, S.M.; Li, Z.; Summanen, P.H.; Downes, J.; Thames, G.; Corbett, K.; Dowd, S.; Krak, M.; Heber, D. Xylooligosaccharide increases bifidobacteria but not lactobacilli in human gut microbiota. Food Funct. 2014, 5, 436–445. [Google Scholar] [CrossRef]
- Yang, J.; Summanen, P.H.; Henning, S.M.; Hsu, M.; Lam, H.; Huang, J.; Tseng, C.H.; Dowd, S.E.; Finegold, S.M.; Heber, D.; et al. Xylooligosaccharide supplementation alters gut bacteria in both healthy and prediabetic adults: A pilot study. Front. Physiol. 2015, 6, 216. [Google Scholar] [CrossRef]
- Sheu, W.H.; Lee, I.T.; Chen, W.; Chan, Y.C. Effects of xylooligosaccharides in type 2 diabetes mellitus. J. Nutr. Sci. Vitaminol. 2008, 54, 396–401. [Google Scholar] [CrossRef]
- Li, F.; Li, Q.; Zhang, Y.; Zhou, X.; Yi, R.; Zhao, X. Effects of Xylooligosaccharides on Lipid Metabolism, Inflammation, and Gut Microbiota in C57BL/6J Mice Fed a High-Fat Diet. Front. Pharmacol. 2021, 12, 791614. [Google Scholar] [CrossRef]
- Chen, H.H.; Chen, Y.K.; Chang, H.C.; Lin, S.Y. Immunomodulatory Effects of Xylooligosaccharides. Food Sci. Technol. Res. 2012, 18, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Fei, Y.; Wang, Y.; Pang, Y.; Wang, W.; Zhu, D.; Xie, M.; Lan, S.; Wang, Z. Xylooligosaccharide Modulates Gut Microbiota and Alleviates Colonic Inflammation Caused by High Fat Diet Induced Obesity. Front. Physiol. 2019, 10, 1601. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.H.; Frokiaer, H.; Christensen, A.G.; Bergstrom, A.; Licht, T.R.; Hansen, A.K.; Metzdorff, S.B. Dietary xylooligosaccharide downregulates IFN-gamma and the low-grade inflammatory cytokine IL-1beta systemically in mice. J. Nutr. 2013, 143, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Hintikka, J.; Lensu, S.; Mäkinen, E.; Karvinen, S.; Honkanen, M.; Lindén, J.; Garrels, T.; Pekkala, S.; Lahti, L. Xylo-Oligosaccharides in Prevention of Hepatic Steatosis and Adipose Tissue Inflammation: Associating Taxonomic and Metabolomic Patterns in Fecal Microbiomes with Biclustering. Int. J. Environ. Res. Public Health 2021, 18, 4049. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Ohba, H.; Sakaki, T.; Takamine, K.; Kamino, Y.; Moriwaki, S.; Bakalova, R.; Uemura, Y.; Hatate, Y. Hot-compressed-water decomposed products from bamboo manifest a selective cytotoxicity against acute lymphoblastic leukemia cells. Toxicol. Vitr. 2004, 18, 765–771. [Google Scholar] [CrossRef]
- Maeda, R.; Ida, T.; Ihara, H.; Sakamoto, T. Induction of Apoptosis in MCF-7 Cells by β-1,3-Xylooligosaccharides Prepared fromCaulerpa lentillifera. Biosci. Biotechnol. Biochem. 2014, 76, 1032–1034. [Google Scholar] [CrossRef]
- Ghosh, A.; Chandra, A.; Dhar, A.; Shukla, P.; Baishya, D. Multi-efficient thermostable endoxylanase from Bacillus velezensis AG20 and its production of xylooligosaccharides as efficient prebiotics with anticancer activity. Process Biochem. 2021, 109, 59–71. [Google Scholar] [CrossRef]
- Hsu, C.-K.; Liao, J.-W.; Chung, Y.-C.; Hsieh, C.-P.; Chan, Y.-C. Xylooligosaccharides and Fructooligosaccharides Affect the Intestinal Microbiota and Precancerous Colonic Lesion Development in Rats. J. Nutr. 2004, 134, 1523–1528. [Google Scholar] [CrossRef]
- Aachary, A.A.; Gobinath, D.; Srinivasan, K.; Prapulla, S.G. Protective effect of xylooligosaccharides from corncob on 1,2-dimethylhydrazine induced colon cancer in rats. Bioact. Carbohydr. Diet. Fibre 2015, 5, 146–152. [Google Scholar] [CrossRef]
- Ávila, P.F.; Martins, M.; de Almeida Costa, F.A.; Goldbeck, R. Xylooligosaccharides production by commercial enzyme mixture from agricultural wastes and their prebiotic and antioxidant potential. Bioact. Carbohydr. Diet. Fibre 2020, 24, 100234. [Google Scholar] [CrossRef]
- Synytsya, A. Fourier transform Raman and infrared spectroscopy of pectins. Carbohydr. Polym. 2003, 54, 97–106. [Google Scholar] [CrossRef]
- Yuan, T.-Q.; Xu, F.; He, J.; Sun, R.-C. Structural and physico-chemical characterization of hemicelluloses from ultrasound-assisted extractions of partially delignified fast-growing poplar wood through organic solvent and alkaline solutions. Biotechnol. Adv. 2010, 28, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Bernardino-Nicanor, A.; Acosta-García, G.; Güemes-Vera, N.; Montañez-Soto, J.L.; de los Ángeles Vivar-Vera, M.; González-Cruz, L. Fourier transform infrared and Raman spectroscopic study of the effect of the thermal treatment and extraction methods on the characteristics of ayocote bean starches. J. Food Sci. Technol. 2016, 54, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Kacuráková, M. FT-IR study of plant cell wall model compounds: Pectic polysaccharides and hemicelluloses. Carbohydr. Polym. 2000, 43, 195–203. [Google Scholar] [CrossRef]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio-Protocol 2019, 9, e3128. [Google Scholar] [CrossRef]
- Okamoto, M.; Hirai, H.; Taniguchi, K.; Shimura, K.; Inaba, T.; Shimazaki, C.; Taniwaki, M.; Imanishi, J. Toll-like Receptors (TLRs) are expressed by myeloid leukaemia cell lines, but fail to trigger differentiation in response to the respective TLR ligands. Br. J. Haematol. 2009, 147, 585–587. [Google Scholar] [CrossRef]
- Zhong, X.; Liu, M.; Yao, W.; Du, K.; He, M.; Jin, X.; Jiao, L.; Ma, G.; Wei, B.; Wei, M. Epigallocatechin-3-Gallate Attenuates Microglial Inflammation and Neurotoxicity by Suppressing the Activation of Canonical and Noncanonical Inflammasome via TLR4/NF-κB Pathway. Mol. Nutr. Food Res. 2019, 63, 1801230. [Google Scholar] [CrossRef]
- Desideri, E.; Ciccarone, F.; Ciriolo, M.R. Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy. Nutrients 2019, 11, 1926. [Google Scholar] [CrossRef]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1926. [Google Scholar] [CrossRef]
- Ferreira de Freitas, R.; Schapira, M. A systematic analysis of atomic protein–ligand interactions in the PDB. MedChemComm 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Xu, Y.; Yu, S. Co-production of functional xylooligosaccharides and fermentable sugars from corncob with effective acetic acid prehydrolysis. Bioresour. Technol. 2017, 234, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Morais de Carvalho, D.; Martínez-Abad, A.; Evtuguin, D.V.; Colodette, J.L.; Lindström, M.E.; Vilaplana, F.; Sevastyanova, O. Isolation and characterization of acetylated glucuronoarabinoxylan from sugarcane bagasse and straw. Carbohydr. Polym. 2017, 156, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zeng, X.; Qiu, J.; Du, J.; Cao, X.; Tang, X.; Sun, Y.; Li, S.; Lei, T.; Liu, S.; et al. Spray-dried xylooligosaccharides carried by gum Arabic. Ind. Crops Prod. 2019, 135, 330–343. [Google Scholar] [CrossRef]
- Yoshino, K.; Higashi, N.; Koga, K. Inhibitory effects of acidic xylooligosaccharide on stress-induced gastric inflammation in mice. Shokuhin Eiseigaku Zasshi. J. Food Hyg. Soc. Jpn. 2006, 47, 284–287. [Google Scholar] [CrossRef]
- Gobinath, D.; Madhu, A.N.; Prashant, G.; Srinivasan, K.; Prapulla, S.G. Beneficial effect of xylo-oligosaccharides and fructo-oligosaccharides in streptozotocin-induced diabetic rats. Br. J. Nutr. 2010, 104, 40–47. [Google Scholar] [CrossRef]
- Cochet, F.; Peri, F. The Role of Carbohydrates in the Lipopolysaccharide (LPS)/Toll-Like Receptor 4 (TLR4) Signalling. Int. J. Mol. Sci. 2017, 18, 2318. [Google Scholar] [CrossRef]
- Fatima, N.; Akhtar, T.; Sheikh, N. Prebiotics: A Novel Approach to Treat Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2017, 2017, 6238106. [Google Scholar] [CrossRef]
- Javaid, N.; Choi, S. Toll-like Receptors from the Perspective of Cancer Treatment. Cancers 2020, 12, 297. [Google Scholar] [CrossRef]
- Fernández-Lainez, C.; Akkerman, R.; Oerlemans, M.M.P.; Logtenberg, M.J.; Schols, H.A.; Silva-Lagos, L.A.; López-Velázquez, G.; de Vos, P. β(2→6)-Type fructans attenuate proinflammatory responses in a structure dependent fashion via Toll-like receptors. Carbohydr. Polym. 2022, 277, 118893. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, X.; Bao, X.; Xiao, W.; Chen, G. Toll-like receptor 4 (TLR4) inhibitors: Current research and prospective. Eur. J. Med. Chem. 2022, 235, 114291. [Google Scholar] [CrossRef]
- Hsu, R.Y.C.; Chan, C.H.F.; Spicer, J.D.; Rousseau, M.C.; Giannias, B.; Rousseau, S.; Ferri, L.E. LPS-Induced TLR4 Signaling in Human Colorectal Cancer Cells Increases β1 Integrin-Mediated Cell Adhesion and Liver Metastasis. Cancer Res. 2011, 71, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Guillot, L.; Medjane, S.; Le-Barillec, K.; Balloy, V.; Danel, C.; Chignard, M.; Si-Tahar, M. Response of Human Pulmonary Epithelial Cells to Lipopolysaccharide Involves Toll-like Receptor 4 (TLR4)-dependent Signaling Pathways. J. Biol. Chem. 2004, 279, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Du, S.; Du, Y.; Ren, J.; Ying, G.; Yan, Z. Glutathione reductase mediates drug resistance in glioblastoma cells by regulating redox homeostasis. J. Neurochem. 2018, 144, 93–104. [Google Scholar] [CrossRef] [PubMed]
- DuBois, M.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.A.; Smith, F. Colorimetric Method for Determination of Sugars and Related Substances. Anal. Chem. 2002, 28, 350–356. [Google Scholar] [CrossRef]
- Blumenkrantz, N.; Asboe-Hansen, G. New method for quantitative determination of uronic acids. Anal. Biochem. 1973, 54, 484–489. [Google Scholar] [CrossRef]
- Singleton, V.L.; Rossi, J.A.J. Colorimetry of total phenolics with phosphomolybdic-phosphotungstic acid reagents. Am. J. Enol. Vitic. 1965, 16, 144–158. [Google Scholar]
- McComb, E.A.; McCready, R.M. Determination of Acetyl in Pectin and in Acetylated Carbohydrate Polymers. Anal. Chem. 2002, 29, 819–821. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and Validation of an Improved Oxygen Radical Absorbance Capacity Assay Using Fluorescein as the Fluorescent Probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Teneva, D.; Pencheva, D.; Petrova, A.; Ognyanov, M.; Georgiev, Y.; Denev, P. Addition of Medicinal Plants Increases Antioxidant Activity, Color, and Anthocyanin Stability of Black Chokeberry (Aronia melanocarpa) Functional Beverages. Plants 2022, 11, 243. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Flanagan, J.; Deemer, E.K.; Prior, R.L.; Huang, D. Novel Fluorometric Assay for Hydroxyl Radical Prevention Capacity Using Fluorescein as the Probe. J. Agric. Food Chem. 2002, 50, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, J.M.; Armstrong, L.S.; Martinez, A.O. A rapid and simple MTT-based spectrophotometric assay for determining drug sensitivity in monolayer cultures. J. Tissue Cult. Methods 1988, 11, 15–17. [Google Scholar] [CrossRef]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Marques, S.M.; Esteves da Silva, J.C.G. Firefly bioluminescence: A mechanistic approach of luciferase catalyzed reactions. IUBMB Life 2009, 61, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 243–250. [Google Scholar]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P.; et al. IEDB-AR: Immune epitope database—analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [PubMed]
- Temml, V.; Kaserer, T.; Kutil, Z.; Landa, P.; Vanek, T.; Schuster, D. Pharmacophore modeling for COX-1 and -2 inhibitors with LigandScout in comparison to Discovery Studio. Future Med. Chem. 2014, 6, 1869–1881. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. UCSF Chimera, MODELLER, and IMP: An integrated modeling system. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Values |
|---|---|
| Total carbohydrate content [%, w/w] | 97.3 ± 2.4 |
| Total uronic acid content [%, w/w] | <1 |
| Monosaccharide composition [%, w/w] | |
| Xylose | 59.4 ± 2.9 |
| Degree of acetylation [mol%] 1 (Acetyl content [%, w/w]) | 3.3 ± 0.1 (0.6 ± 0.01) |
| Glucose | 9.7 ± 1.1 |
| Total proteins [%, w/w] | n.f. 2 |
| Total phenolics [%, w/w] | 0.2 ± 0.01 |
| ORAC 3 [μmol TE 4/g] | 1150.2 ± 32.8 |
| HORAC 5 [μmol GAE 6/g] | 303.1 ± 9.7 |
| A549 | HT-29 | U-937 | MRC-5 | |
|---|---|---|---|---|
| Mean IC50 (μ g/mL) | 143.3 ± 7.1 *** | 51.8 ± 0.4 *** | 150 ± 9.6 *** | 367.3 ± 9.3 |
| Selectivity index | 2.6 | 7.1 | 2.5 | NA |
| Xylobiose (kcal/mol) | Xylotriose (kcal/mol) | Xylotetraose (kcal/mol) | Xylopentaose (kcal/mol) | Xylohexaose (kcal/mol) | Xyloheptaose (kcal/mol) | |
|---|---|---|---|---|---|---|
| TLR4 | −6.4 | −6.7 | −7.37 | −8.1 | −8.3 | −7.6 |
| huGR | −7.7 | −9.7 | −9.7 | −11.0 | −11.7 | −11.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batsalova, T.; Georgiev, Y.; Moten, D.; Teneva, I.; Dzhambazov, B. Natural Xylooligosaccharides Exert Antitumor Activity via Modulation of Cellular Antioxidant State and TLR4. Int. J. Mol. Sci. 2022, 23, 10430. https://doi.org/10.3390/ijms231810430
Batsalova T, Georgiev Y, Moten D, Teneva I, Dzhambazov B. Natural Xylooligosaccharides Exert Antitumor Activity via Modulation of Cellular Antioxidant State and TLR4. International Journal of Molecular Sciences. 2022; 23(18):10430. https://doi.org/10.3390/ijms231810430
Chicago/Turabian StyleBatsalova, Tsvetelina, Yordan Georgiev, Dzhemal Moten, Ivanka Teneva, and Balik Dzhambazov. 2022. "Natural Xylooligosaccharides Exert Antitumor Activity via Modulation of Cellular Antioxidant State and TLR4" International Journal of Molecular Sciences 23, no. 18: 10430. https://doi.org/10.3390/ijms231810430
APA StyleBatsalova, T., Georgiev, Y., Moten, D., Teneva, I., & Dzhambazov, B. (2022). Natural Xylooligosaccharides Exert Antitumor Activity via Modulation of Cellular Antioxidant State and TLR4. International Journal of Molecular Sciences, 23(18), 10430. https://doi.org/10.3390/ijms231810430