2.1. Erlotinib Conjugates and Cellular Uptake
The current functional study involved examples of erlotinib chlorin conjugates described previously [
11] and which are depicted in
Figure 1. The original designations of these compounds were retained to facilitate cross-comparison of data. Sets of erlotinib conjugates had been synthesized to evaluate the functional impact of several specific structural features: flexible or rigid linker (PS 5 vs. PS 8); attachment of erlotinib to position 17
2 or position 3
1 (PS 8 vs. PS 10); contribution of an acidic charge at position 17
2 to cellular retention (PS 10 vs. PS 11); attachment of erlotinib to the meta or para position of the rigid benzyloxy linker (PS 10 vs. PS 16), a chiral methyl group at position 3
1 of pyropheophorbide (PS 10 vs. PS 19); increase to three erlotinib moieties (PS 8 vs. PS 27); and attachment of erlotinib to position 13 of chlorin e6 with short or long linker structure (PS 38 vs. PS 43). The initial characterization of additional compounds had already ruled out as ineffective those PSs with erlotinib conjugated to position 20 via benzoic acid linker of pyropheophorbide (PS 23) or to the imide ring of purpurinimide (PS 29 and PS 31).
Following information regarding cellular activity had been obtained [
11]. Erlotinib attached by rigid linker reduced cellular uptake. Uptake was further lowered when erlotinib was placed into para position of the benzyloxy linker. Retention was also attenuated when pyropheophorbide is missing the chiral methyl group at position 3
1. The intracellular sites of deposition for every PS with a single erlotinib moiety appeared to be the same as for non-conjugated HPPH. The highest level of cellular uptake, but coupled with reduced tumor cell specificity, was determined for PS 5. In contrast, PS 10, although taken up at moderate level, demonstrated highest differential retention by a subset of tumor cells. Uptake and photodynamic activities measured in cell culture systems could be confirmed in vivo using xenografts of established human cancer cell lines. However, none of the preliminary cell studies provided evidence for a specific involvement of an erlotinib-sensitive and EGFR-dependent action. The current study addresses this issue.
To be effective as substrate for binding to EGFR, as well as to act as inhibitor of the EGFR kinase, erlotinib conjugates must gain access to the cytoplasmic domain of the EGFR protein. Hence, excluded are those compounds that are endocytosed and deposited to lysosomes, such as PS 27. To visualize the mode of cellular entry and intracellular localization of the erlotinib conjugates under investigation, we employed reconstituted co-cultures of head and neck (HN) tumor epithelial cells (T-EC) and tumor-associate fibroblasts (T-Fb) (
Figure 2; example of tongue tumor-derived HNT1 cells combined with carboxyfluorescein succinimidyl ester (CFSE)-tagged HN tumor stromal cells). Incubations for 30 min demonstrated the immediate and high-level entry of HPPH by diffusion into both cell types and incubation for 24 h yielded maximal level of HPPH accumulation. The subcellular site of deposition is mitochondria/ER, which is clearly distinct from lysosomal compartment (
Figure 3A, left 2 panels). The relative retention of HPPH during the subsequent 48 h chase period defined the cell type specificity (
Figure 2).
Compared to HPPH, the erlotinib pyropheophorbide derivatives showed a 4-fold (PS 10, PS 19) to 20-fold (PS 16) lower initial uptake coupled with a reduced accumulation over the subsequent hours (
Figure 2). The comparison also indicated the structure-dependent 48 h retention by the cell types with PS 10 as most prominently retained substrate for T-EC (
Figure 2). Despite the lower uptake, erlotinib conjugates demonstrated a pattern of intracellular deposition equivalent to HPPH that, as shown for PS 10 and PS 11 in HN-143 T-EC (tongue cancer), is congruent with that of mitotracker (
Figure 3A, right 2 panels). Comparable results have been obtained for the other erlotinib conjugates (except PS 27, which fails to diffuse into cells).
These co-culture experiments suggest that the erlotinib conjugates potentially encounter the intracellular domain of the plasma membrane EGFR during the internalization process. This encounter appears to be transient with the subsequent sequestration of the PSs at distant mitochondria/ER. Whether the inferred encounter after 30 min or after 24 h treatment is sufficient to alter EGFR kinase activity needed to be determined.
Indirect evidence for a failure of an interaction between erlotinib conjugates and EGFR as part of the uptake process was obtained when a maxilla cancer (HN-124) was identified whose tumor cells exhibited a rare phenotype of extremely low-level expression of EGFR protein and activity (
Figure 3C). Despite of this deficiency, HN-124 T-EC did take up erlotinib-conjugates, such as PS 10, even more efficiently than HNT1 cells (
Figure 3B) that express >100-fold higher level of EGFR (
Figure 3C).
2.2. EGFR Activity in Cells Treated with Erlotinib Pyropheophorbide Conjugates
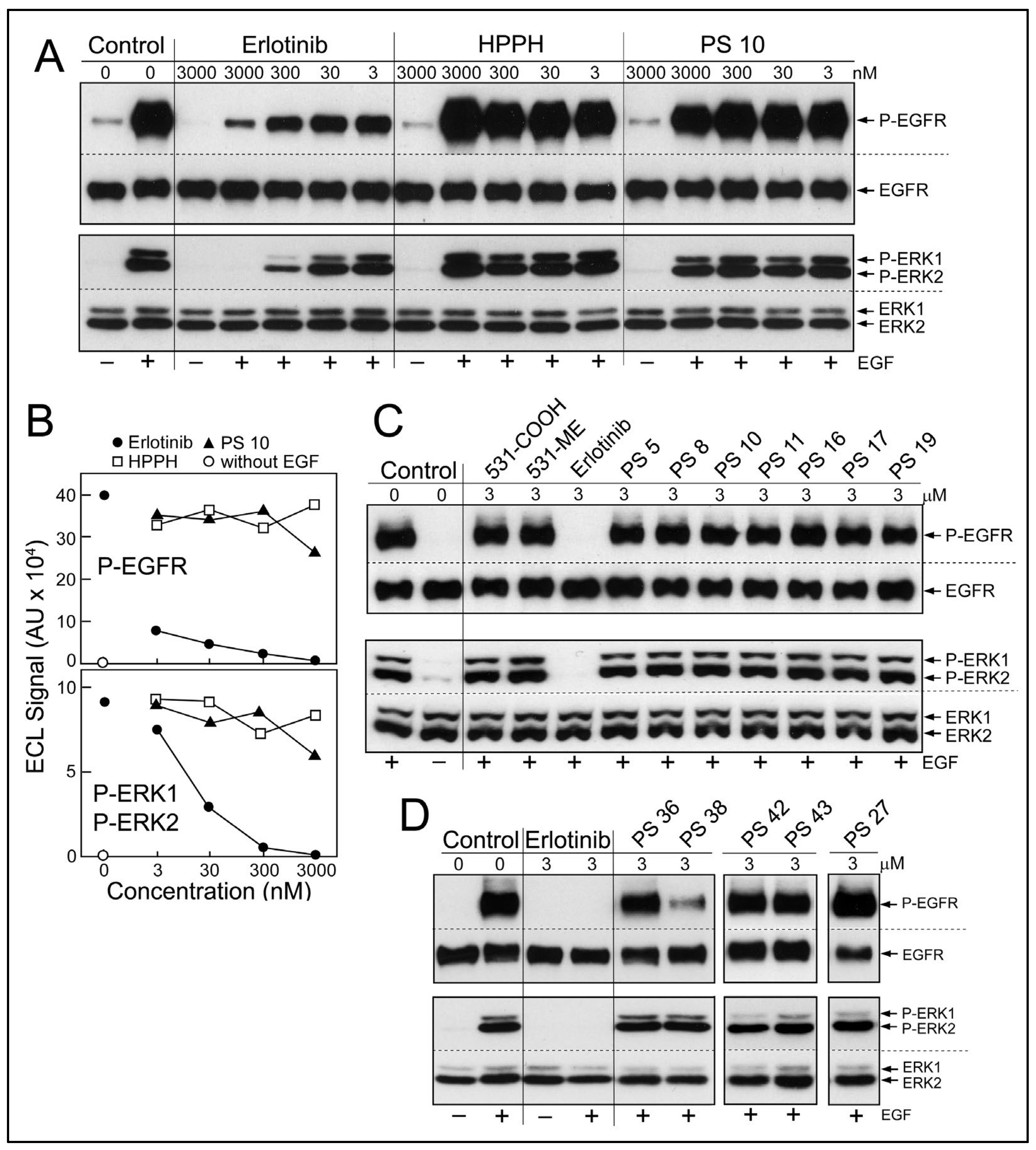
Diffusion of erlotinib-PS conjugates into cancer cells predicts the potential encounter of these with the cytoplasmic domain of EGFR and inhibition of its kinase function. To test this prediction, a cell-based assay of EGFR activity was applied (
Figure 4). Cultured HN tumor cells with high level expression of EGFR protein and kinase activity, such as HNT1 cells, were pretreated with erlotinib or erlotinib-pyropheophorbides for 15 min in serum-free medium (
Figure 4A,B) or for 24 h in medium containing 10% FBS (
Figure 4C,D), followed by addition of EGF into the same medium and continued incubation for 15 min. Two pretreatment periods were chosen considering the different uptake kinetics and accumulation levels of the erlotinib-conjugates (
Figure 2). EGF-dependent stimulation of EGFR kinase activity was evaluated by the autophosphorylation of EGFR and signal transduction toward the ERK1/2 pathway (
Figure 4A). The inhibitory activities of non-conjugated erlotinib indicated an IC50 < 3 nM for P-EGFR and an IC50 ~5 nM for P-ERK1/2 (
Figure 4B); values that are in the range of those determined in other cancer cell types [
12,
13].
Although there were substantial differences in the ability of HN cancer cells to take up erlotinib conjugates [
11], there was no significant reduction of ligand-dependent EGFR activity, even after 24 h incubation (
Figure 4C,D). One exception was PS 38, chlorin e6 with erlotinib attached to position 13 by a benzyloxy linker. PS 38 appreciably reduced the more sensitive read-out of EGFR activity, the autophosphorylation of the receptor protein (
Figure 4D). The related compound PS 43, chlorin e6 with erlotinib attached to the same position 13 but via an ethylene glycol linker, was ineffective as EGFR inhibitor. The cell-based kinase assay was limited to a highest concentration of 3 μM PS, a concentration that exceeded several-fold that of the same PSs used for PDT.
2.3. PS-Mediated Photoreactions
Previous studies on the photosensitizing properties of pyropheophorbides, including HPPH, have indicated a tight correlation between the amount of PS taken up by the cells, as determined by the PS fluorescence, and the light-dependent photoreaction mediated by the PS [
2]. The photoreaction was quantified by the covalent crosslinking of the latent transcription factor STAT3, loss of EGFR and activation of the stress p38 MAP kinase pathway. The same experimental analysis was applied to erlotinib conjugates. To assess the range of cell-specific PS action, T-EC preparations from different HN tumor samples with distinct binding activity for HPPH, were incubated with erlotinib-conjugates and the relationship of PS retention and response to the photoreaction determined.
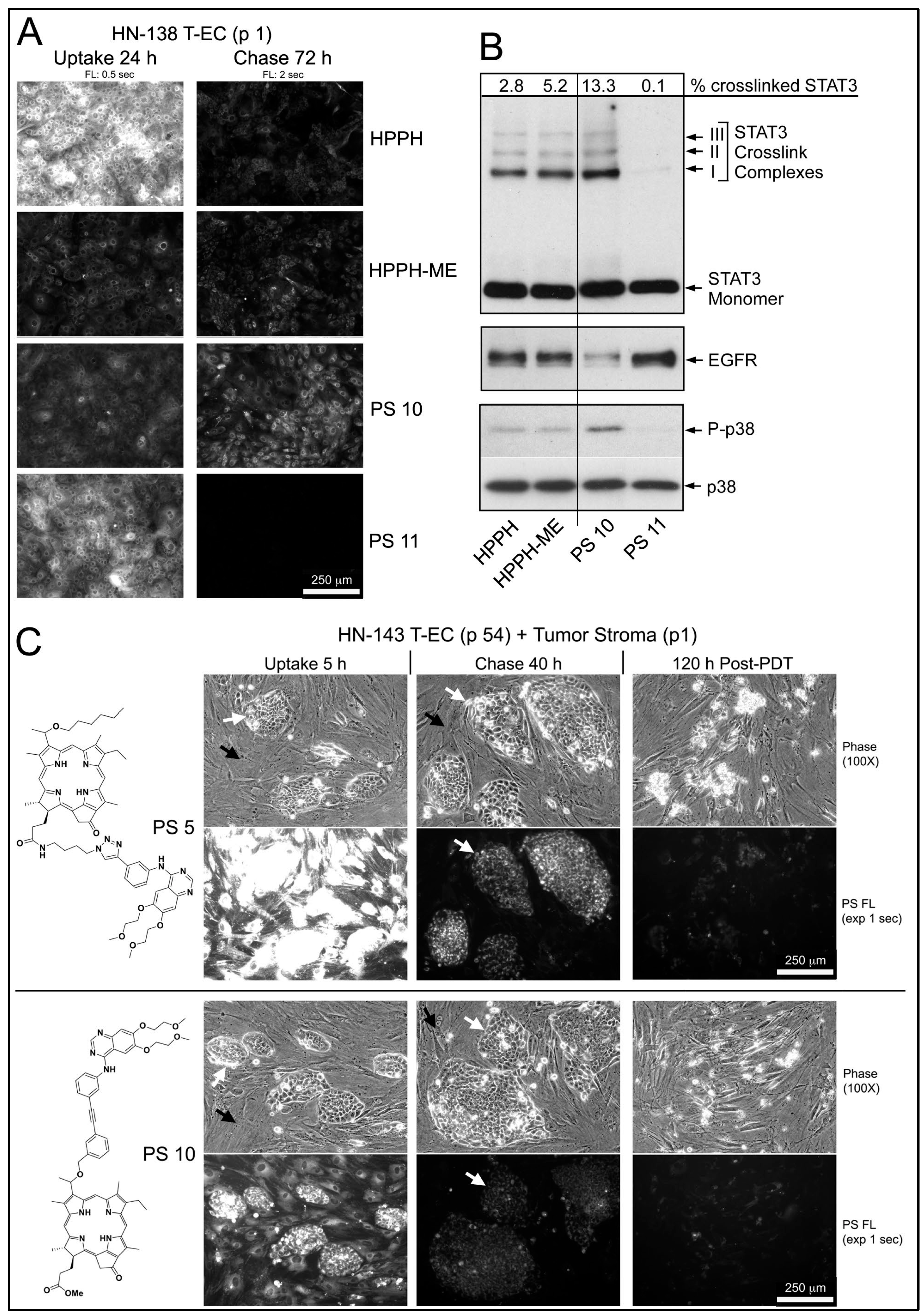
In the example of an early passage of HN-138 T-EC (larynx tumor), which had indicated a low HPPH-retaining activity, the structurally related pair of PS 10 and PS 11 (differing in the presence of a methylester versus a carboxyl group at position 17
2) was compared to HPPH and HPPH-ME (
Figure 5A,B). Although the carboxylated compounds (HPPH and PS 11) were taken up at several-fold higher levels than the corresponding methylester derivatives (HPPH-ME and PS 10), after 72 h chase, only minor fraction remained (
Figure 5A). In contrast, the methylester derivatives showed much lower egress rates with PS 10 as having the highest remaining level. Treatment of these cultures with therapeutic 665-nm light elicited photoreactions, which were proportional to the PS level (
Figure 5B). As reported previously, the relative value for STAT3 dimerization in turn predicts the level of 24 h post-PDT survival of the cultures [
2]. A survey of >100 separate monoytypic T-EC cultures for their lethal response to HPPH-mediated PDT indicated a “LD50” equivalent when ~1% of cellular STAT3 has been converted into dimers [
2]. Complete lethal response was reached with 2–10% STAT3 dimerization. Hence, in the case of the HN-138 T-EC cultures used in
Figure 5A,B, the effectiveness of PDT by the PSs is ranked PS 10 > HPPH-ME > HPPH > PS 11, with the last one yielding minimal, if any lethal reaction.
The application of erlotinib-PS in murine tumor models had indicated an unexpectedly high PDT efficacy for several conjugates. Although the cellular level of these PS did not appreciably exceed that of HPPH, higher lethal cell response and lower tumor recurrence were noted [
11]. The standard analysis of PDT-mediated cell death in tissue culture was limited to the determination of cell viability 24 h after therapeutic light treatment. To assess PDT effect on long-term cell survival and recovery of proliferation, the post-PDT culture period must be extended to several days. Moreover, the influence of cellular interaction within the microenvironment of the tumor tissue needs to be considered. Indeed, growth analyses of many HN T-EC preparations have indicated that proliferation of the tumor cells is enhanced by stromal cell-derived growth factors. Hence, to characterize post-PDT tumor cell survival and growth, we chose to use reconstituted co-cultures of tumor epithelial and stromal cells as test system. The fact that stromal cells preferentially lost HPPH and erlotinib-conjugates (
Figure 2), these cells were expected to survive PDT [
14] and to potentially contribute growth-promoting factors to those tumor cells which escaped lethal PDT.
An example of an effective growth support activity by stromal cells was determined in combination with HN-143 tongue tumor cells (
Figure 5C). Five-day-old co-cultures were incubated for 5 h with PS 5 or PS 10 and chased for 40 h in PS-free medium. Both PSs in the stromal cells and PS 10 in tumor cells were reduced below lethal PDT level, whereas PS 5 in tumor cells was retained at lethal level. Treatment of the co-cultures with 665-nm light followed by 5-day post-PDT incubation in full growth medium resulted, as expected, in the survival of the stromal cells, but with the elimination of all tumor cells (
Figure 5C, right side panels). This seemingly paradox lethal action on PS 10-containing tumor cells, which should have survived at least in part, suggested an unexpected contribution of a PDT-dependent process.
2.4. Release of Erlotinib Activity by Photoreaction
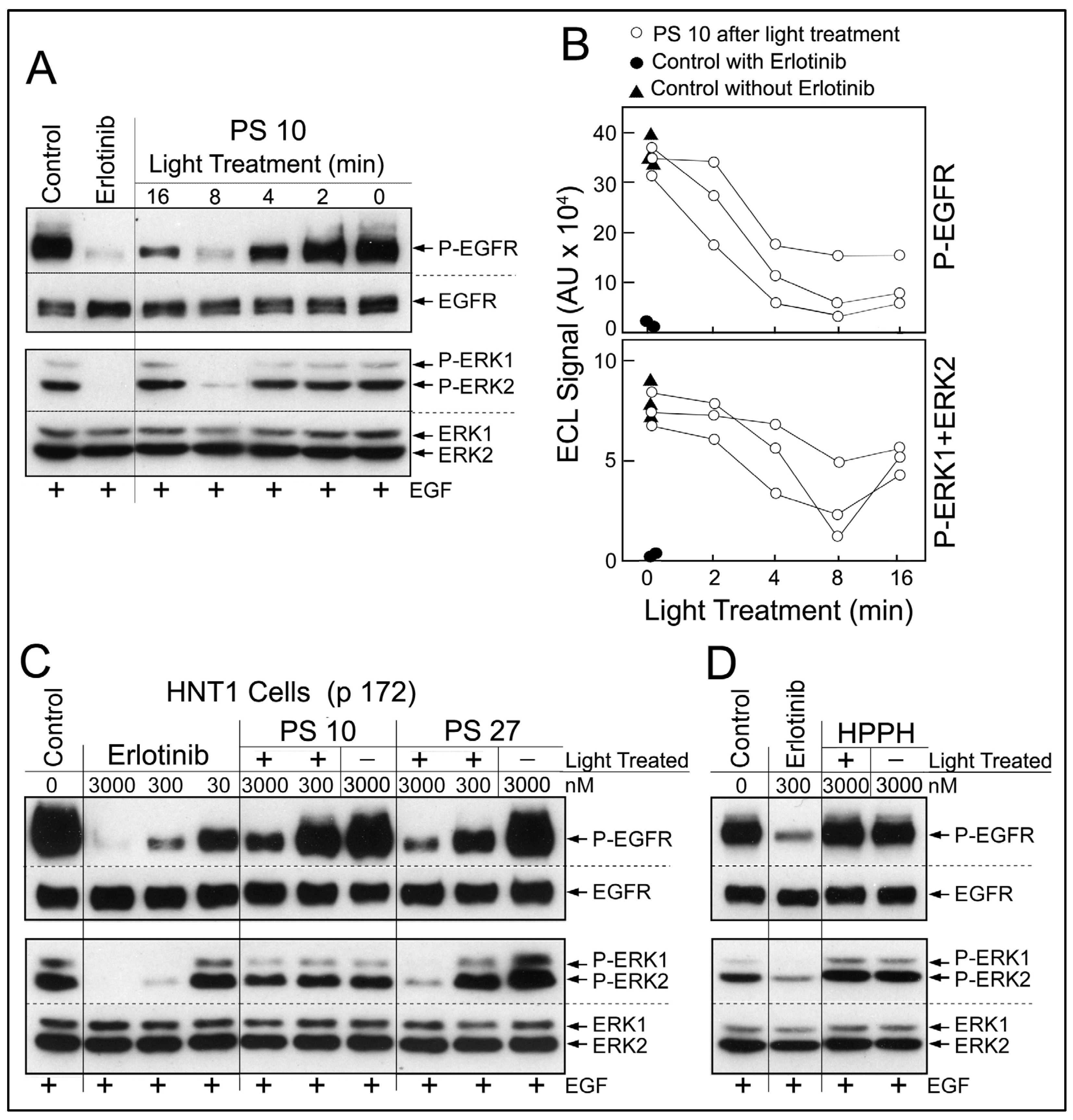
Analysis of the photoreaction mediated by erlotinib conjugates in co-cultures in the presence of serum-containing medium was not amenable to yield conclusive information about growth-modifying components due to the complexity of reaction products. Therefore, we focused on determining the effect of the photoreaction on erlotinib conjugates alone. PS 10 dissolved in methanol was exposed to 660-nm light. Light treatment for 16 min (=5.3 J/cm
2) led to ~40% reduction of PS fluorescence due to photobleaching. Aliquots of the reaction mixture were collected over the course of light treatment. The products were added to the cell-based EGFR kinase assay at a concentration equivalent to 3 μM original PS 10 (
Figure 6A). A light treatment-dependent production of EGFR-inhibitory activity was detected. Maximal inhibitory activity was generated by 8 min light treatment (=2.65 J/cm
2). Prolonged photoreaction led to a reduced recovery of EGFR-inhibitory activity, probably due destructive oxidative processes [
15]. Using the densitometric values for the immunoblot signals for phosphorylated EGFR and ERK, a quantitative estimate of the photochemical release of erlotinib-type activity from PS 10 was obtained (
Figure 6B). Three separate sets of photoreactions yielded comparable time course of inhibitor release, although the relative recovery of inhibitory activity was variable.
Based on testing serially diluted preparations of erlotinib and light-treated PS 10 (
Figure 6C), we estimated a recovery of 1 to 10% of erlotinib-like inhibitory activity from PS 10. Equivalent recovery was obtained with PS 11, but insignificant amount with PS 5 and PS 8. Surprisingly, the photoreaction mediated by PS 27 generated the highest recovery of EGFR-inhibitory activity (
Figure 6C), probably due to the presence of triple erlotinib moieties. We interpret the presence of EGFR-inhibitory activity in light-treated subset of erlotinib conjugates to be due to partial photochemical breakdown of the conjugates releasing erlotinib moieties now effective as EGFR kinase inhibitors. The precise molecular structure of the photoproducts in any of the PS preparations remains to be determined. The photoreaction with non-conjugated HPPH did not result in the production of detectable EGFR-inhibitory activity (
Figure 6D) ruling out reactive components derived from the pyropheophorbide moiety alone.
2.5. Erlotinib PS Conjugate-Derived Photoproducts Attenuate Tumor Cell Proliferation
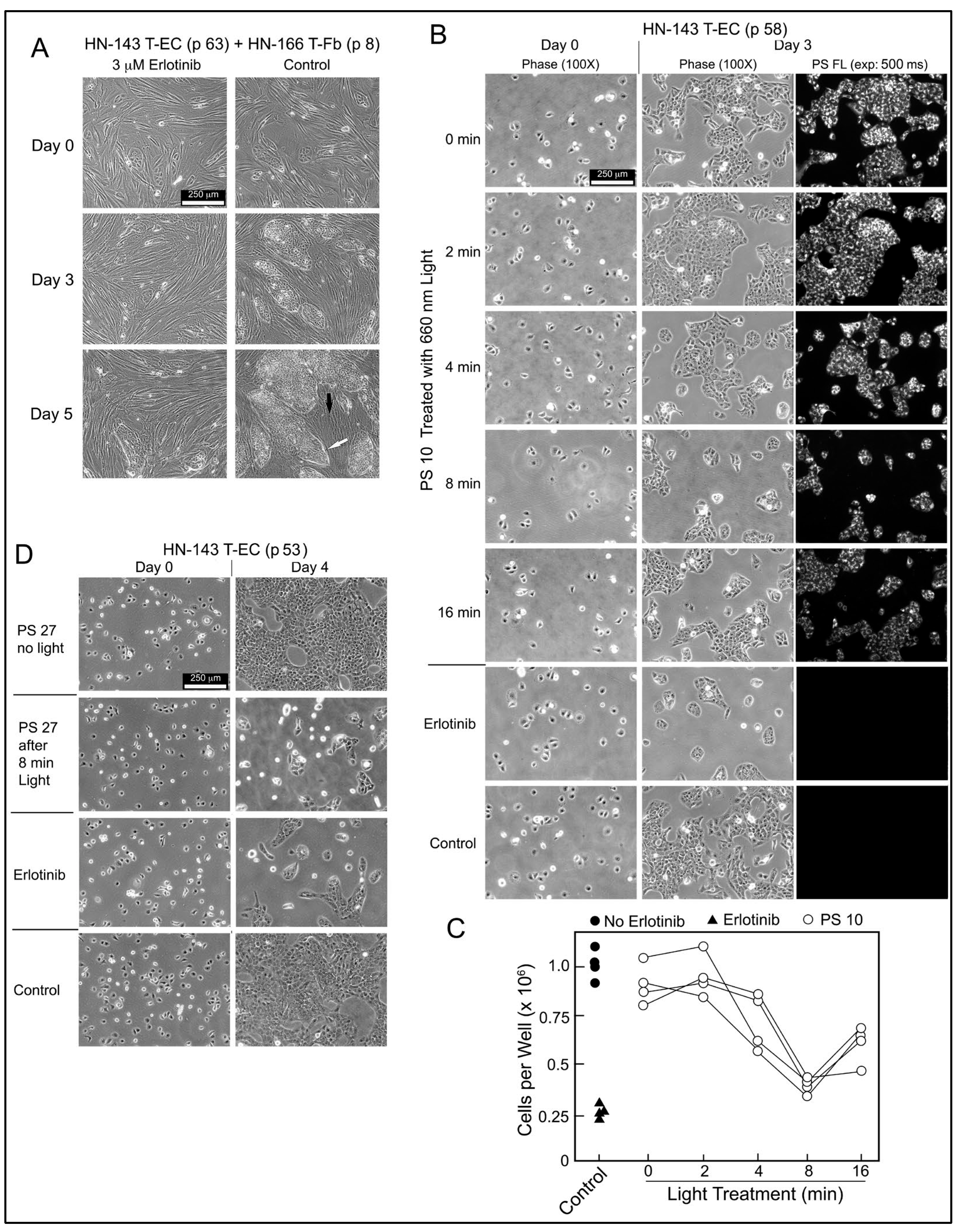
The biological relevance of PS-derived EGFR-inhibitory activity was evaluated using HN-143 T-EC cultures. These tongue cancer cells depend on EGFR signals to proliferate as demonstrated by the drastically reduced growth of HN-143 cells alone or in co-culture with HN tumor stromal cells in the presence of erlotinib (
Figure 7A). Based on the EGFR-inhibitory activity present in light-treated PS 10 preparation (
Figure 6A,B), the same products were applied to HN-143 T-EC cultures and compared to erlotinib (
Figure 7B). After 3 days incubation, the lower cell numbers mirrored the EGFR-inhibitory profile. The cellular level of PS 10 detectable by fluorescence in the cultured cells was proportional to the fluorescence retained by PS 10 after light pretreatment. Four independent series of light-treated PS 10 samples confirmed the time course by which the photoreaction mediated the release of EGFR- and cell growth-inhibitory activity (
Figure 6C and
Figure 7C).
The same cell assay also highlighted the accuracy of testing erlotinib conjugate products by the response to light-treated PS 27 (
Figure 7D). PS 27 has not been considered for further studies because it does not diffuse into cells, is poorly taken up by HN EC, and is primarily deposited into lysosomes. However, it mediates a photoreaction that releases erlotinib-equivalent activity that is now diffusible, inhibits EGFR activity and attenuates proliferation. Hence, despite its uptake into lysosomes, intact PS 27 could assist in reducing recovery of light-treated tumor cells through attenuation of EGFR signaling. The limiting factor would be the amount of PS 27, or of any other erlotinib conjugate, that can reach tumor tissue, and to be retained by cancer cells for mediating a photoreaction sufficiently effective for production of cell damaging reactive oxygen species [
15] and for liberating erlotinib-related products that attenuate localized growth-supporting signaling. Notable is that most of the erlotinib-related activity should be confined to tumor cells. If cells survive the photoreaction, the PS-released erlotinib activity may exert EGFR inhibition and attenuated proliferation. It has already been shown that systemically administered erlotinib in combination with PDT, result in improved PDT outcome, despite the expected action of erlotinib in non-tumor cells [
16,
17,
18].
2.6. In Vivo Distribution of Erlotinib Pyropheophorbides and Uptake by Tumor Tissue
The experimental work with cell culture models has provided information about preference of tumor epithelial cells for the various PSs (
Figure 2), the site of intracellular retention (
Figure 3) and the consequence of light-triggered photoreaction (
Figure 5,
Figure 6 and
Figure 7). When attempting to translate this knowledge to the same tumor cell type but grown in vivo, one recognizes that the key information regarding systemic distribution and the level of uptake of the PSs at local tumor tissue needs to be gained through in situ measurements [
8]. The subsequent assessment of the relationship between cellular concentration of PS in tumor tissue and immediate PDT response in vivo is expected to be much in line with what has been defined in ex vivo models.
The initial characterization of the erlotinib conjugates in murine models of epithelial tumor types had determined the organ distribution [
11]. The method of choice was whole-body imaging of PS fluorescence of infused PS preparations. While these analyses had provided an estimate for the gross anatomical location of the PSs as a function of time following injection, the image resolution was insufficient to inform about cellular and subcellular level of each PS. To gain this information and to quantitatively compare retention of key erlotinib-PS conjugates, we resorted to the imaging of tissue cryosections. The same microscopic detection system was used that we have applied to the analysis of tissue cultures.
Patient-derived bladder cancer (BC-3) xenografts grown in NSG mice were chosen as test model (
Figure 8). BC-3 tumor cells have high level expression of EGFR and form well-defined tumor cell clusters separated by stromal tissue within subcutaneously propagated xenografts. The in vivo properties of three structurally related PSs, PS-531, PS 10 and PS-908, were compared. PS-531 [
6] had served as substrate for the synthesis of PS 10. PS-908 [
10] tested an alternative position of erlotinib in that erlotinib was conjugated via a benzyl linker to position 17
2 as found in PS 8. Organ distribution of the PSs was determined 24 h after intravenous injection by fluorescence microscopy of 10 μm cryosections. All three PSs show uniform and high-level retention in liver with PS-908 reaching the highest concentration. Kidney retained the same PSs in a more heterogenous pattern within tubular cells. BC-3 xenograft on average had level equal to 25% of the liver for PS-531 and PS 10, and 10% of the liver for PS-908. The most relevant information gained by the in vivo experiments as shown in
Figure 8 is that the erlotinib conjugates reached within 24 h a fairly uniform access to the tumor cell clusters, were retained by the tumor cells markedly above the stromal area, and presented subcellular deposition as already determined of tissue culture cells. Few regions within large tumor area showed a reduced level of PS fluorescence at the center and elevated level at periphery, suggesting heterogeneity in intra-tumoral distribution or diffusion of the PSs.
A separate experiment tested the regional distribution of PS 10 in a subcutaneous HNT1 xenograft by using the same treatment and analysis technique as applied for BC3 xenograft-bearing mice. The cryosection of the HNT1 tissue indicated an intra-tumoral distribution of PS 10 with heterogeneity among tumor cell clusters, which was comparable to that seen for BC3 (
Supplementary Figure S1).
The level of PS fluorescence detected in cryosection allows a prediction about the relative effectiveness of the photoreaction and its impact on cell survival. In the case of PS 10 treatment, the release of EGFR-inhibitory activity as a function of the local concentration of PS 10 may appreciably contribute to the post-PDT programing of the surviving tumor cells, activity of the local stromal cells and of inflammatory cells entering post-PDT site. This suggested mechanism may also explain the higher PDT efficacy of PS 10 compared to HPPH detected in the previous in vivo tumor treatment models [
11].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







