
Discovery of Mitophagy Inhibitors with Therapeutic Potential in Different Familial Amyotrophic Lateral Sclerosis Mutations
 , , ,
, , ,  , , and
, , and 
Abstract
:1. Introduction
2. Results
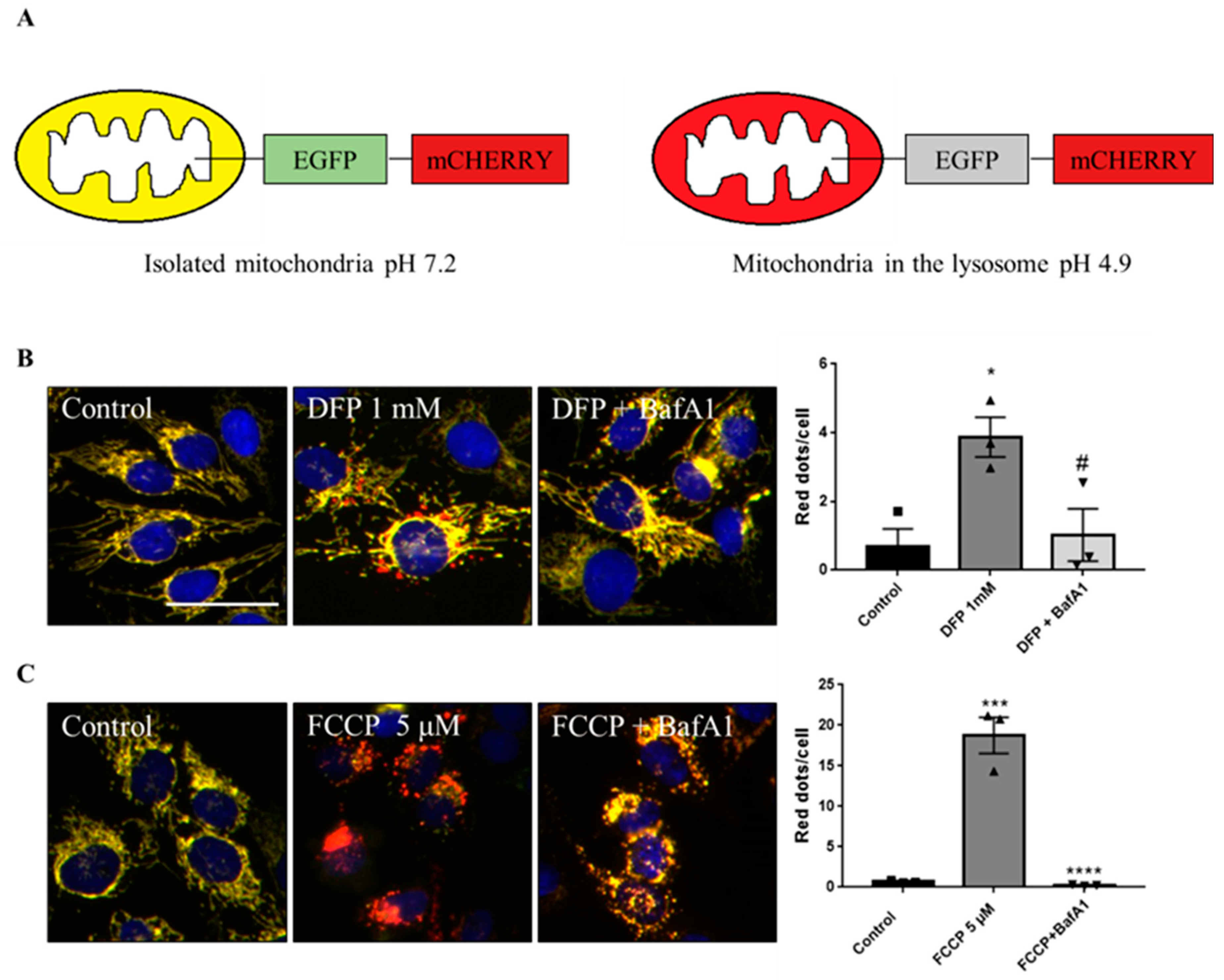
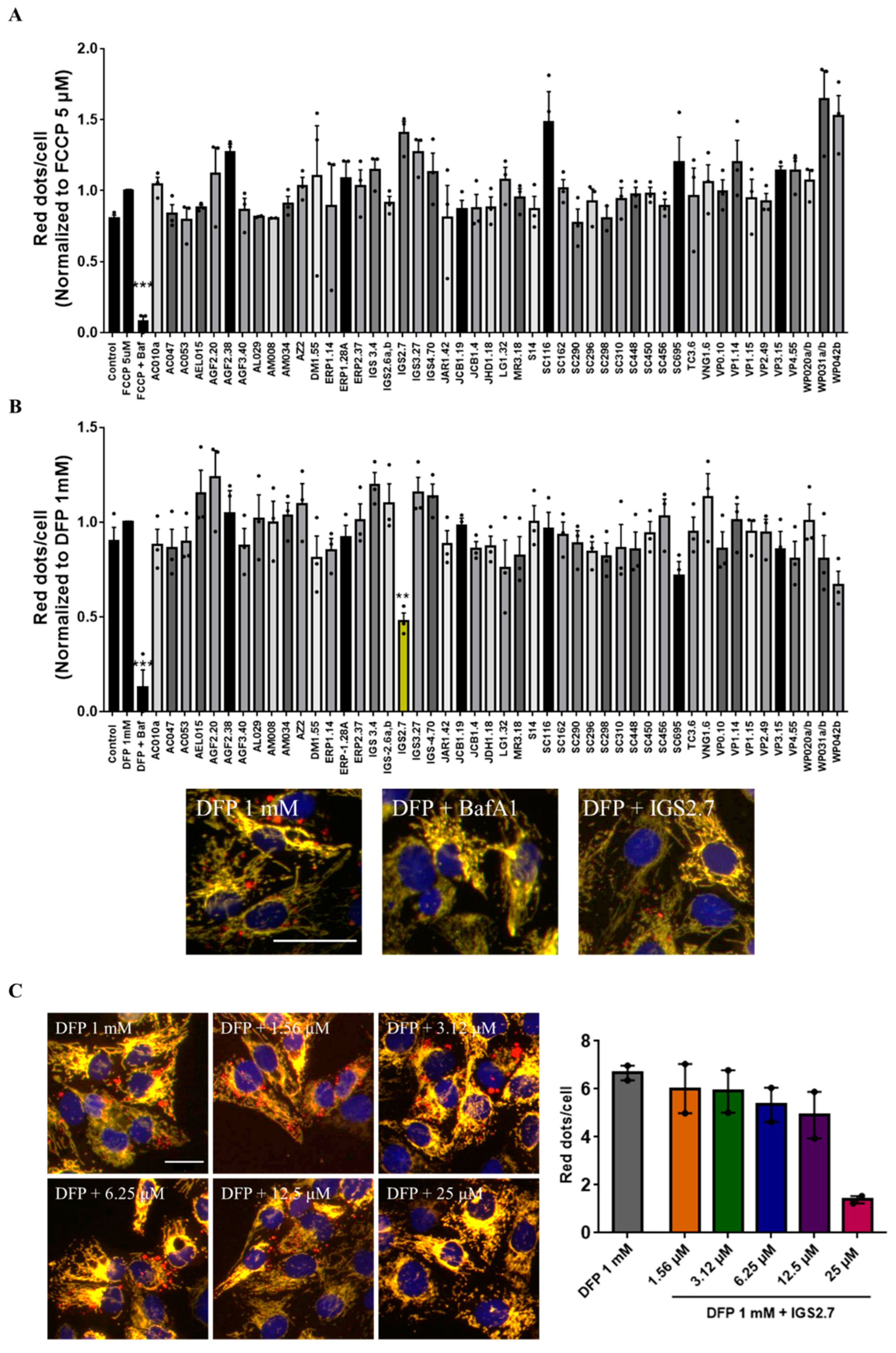
2.1. Phenotypic Screen to Identify Mitophagy Inhibitors
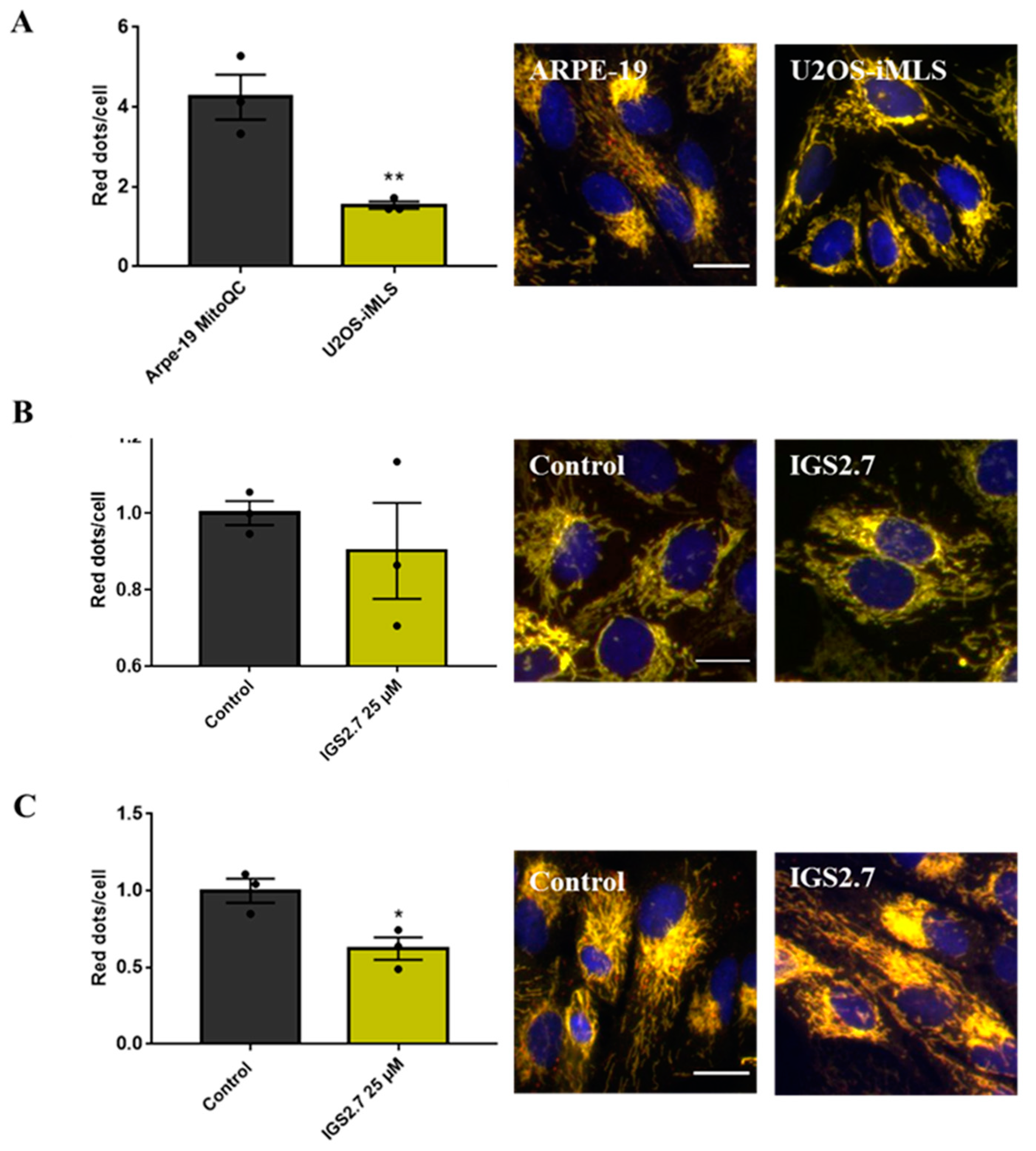
2.2. The Ability of IGS2.7 to Block Basal Mitophagy Is Cell Type Dependent
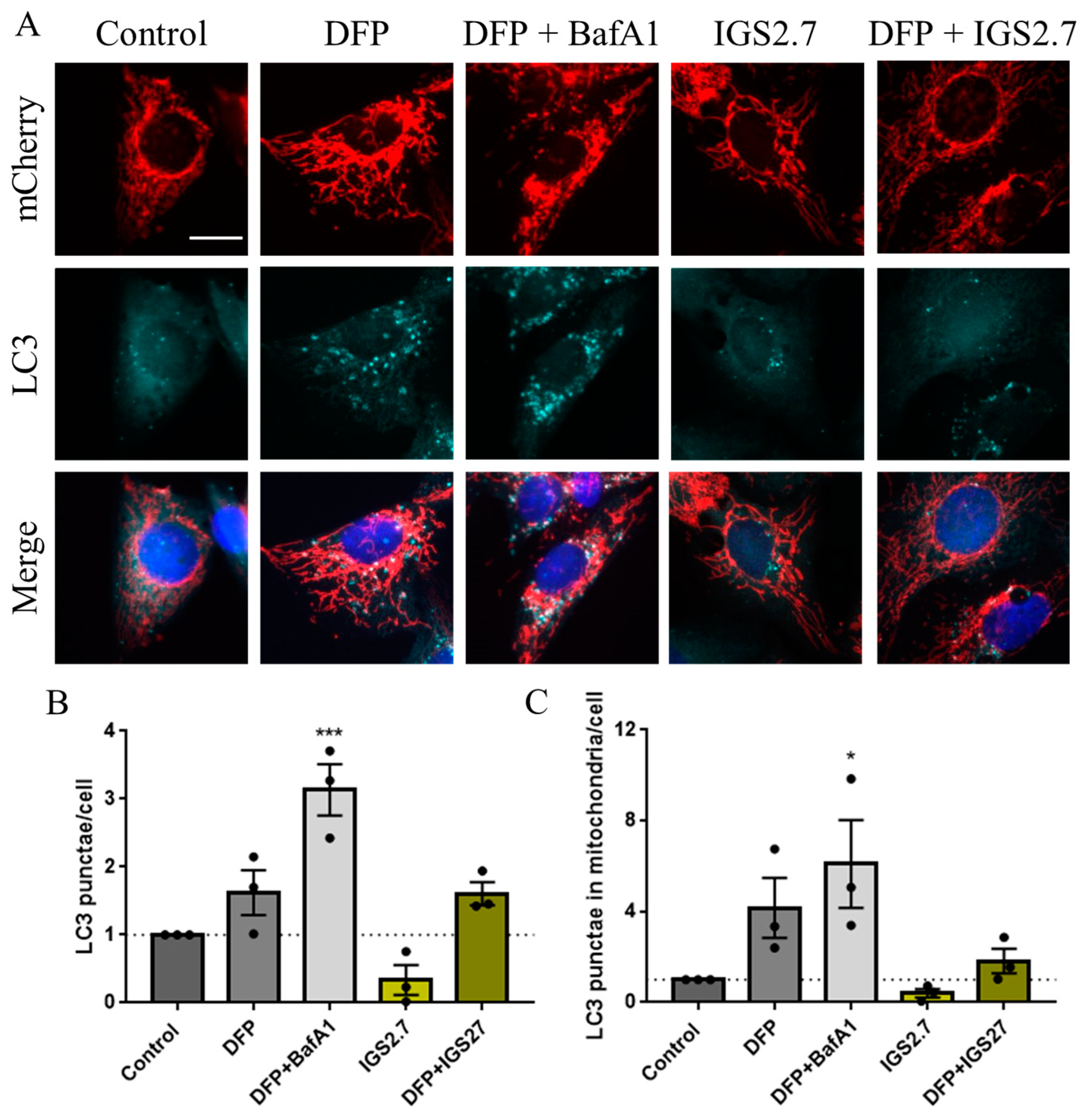
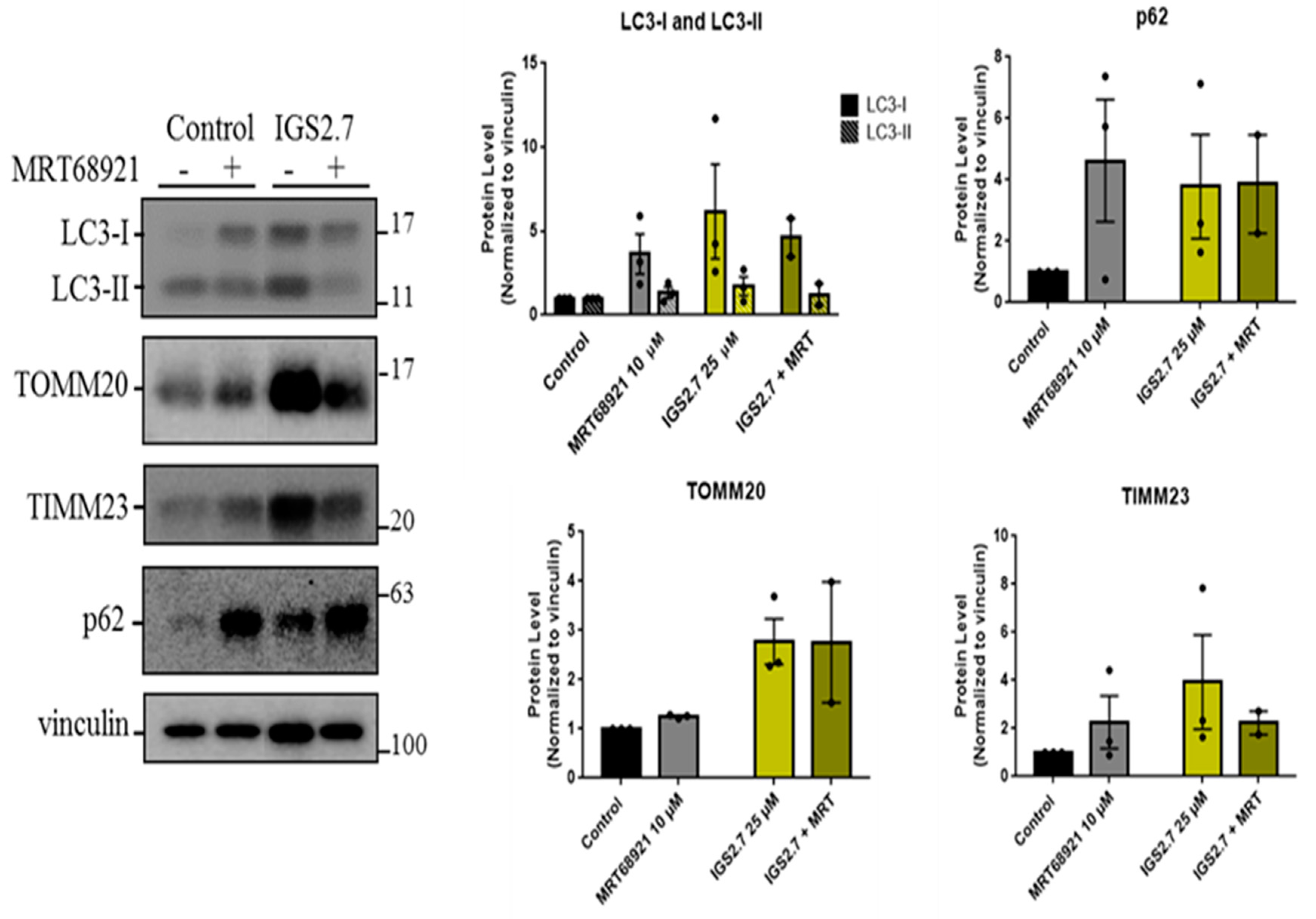
2.3. Potential Mechanism of Action of IGS2.7 in Mitophagy Modulation
2.3.1. IGS2.7 As a Protein Casein Kinase 1 (CK1) Inhibitor
2.3.2. IGS2.7 as an Unc-51 such as Kinase-1 (ULK1) Inhibitor
2.4. Therapeutic Potential of IGS2.7 in ALS Models as Autophagy/Mitophagy Inhibitor
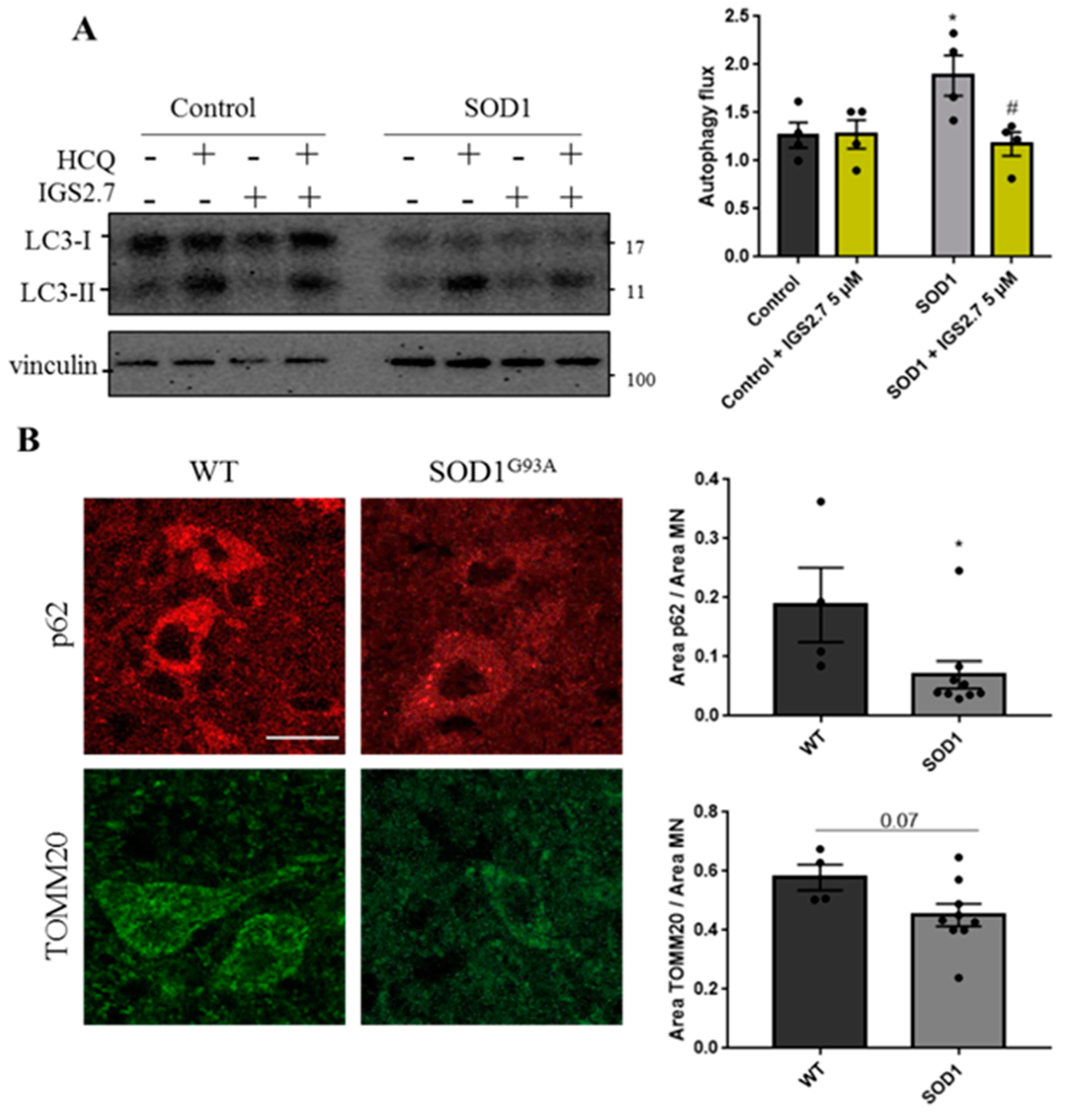
2.4.1. Cellular and Animal SOD1 Model of ALS
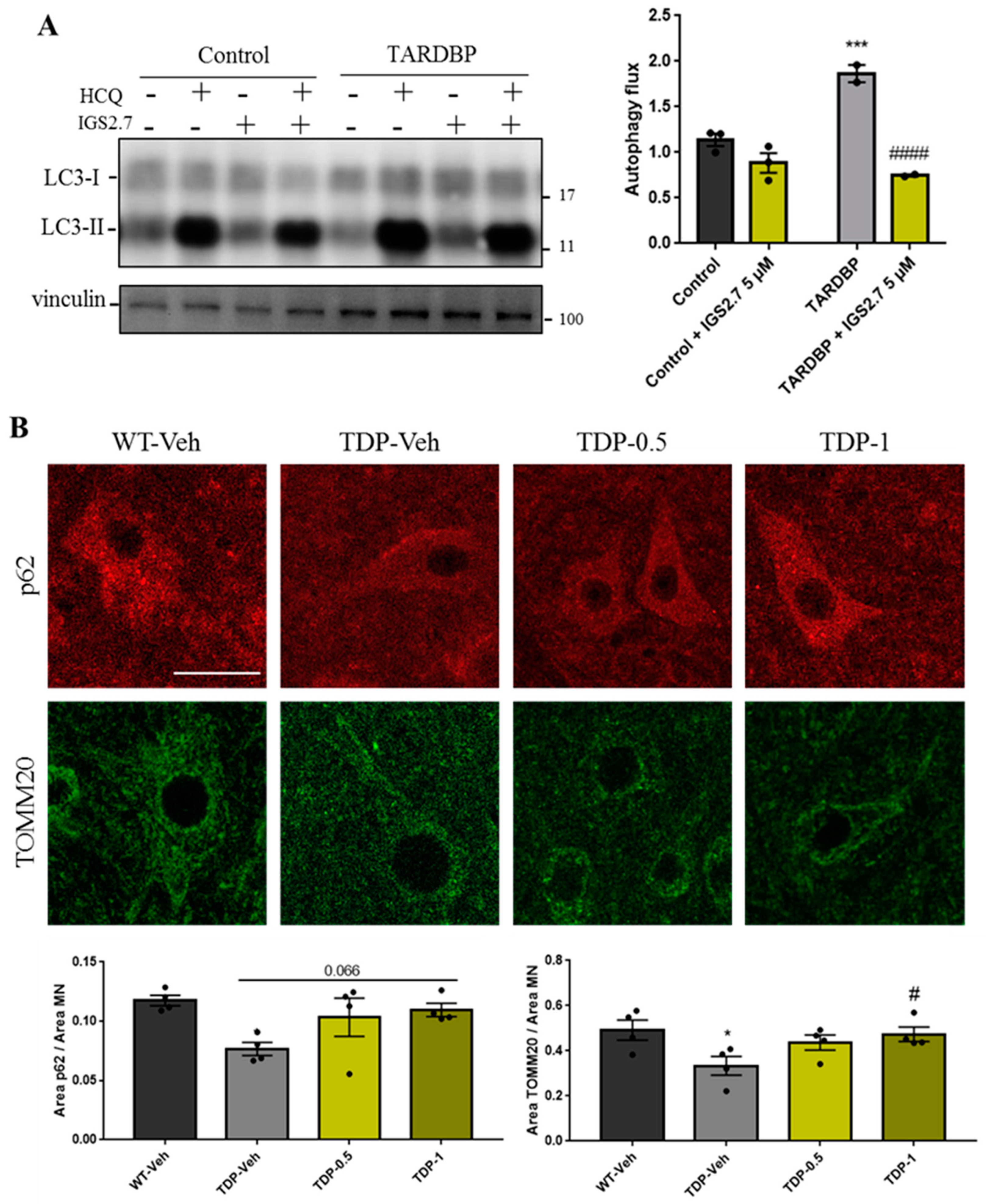
2.4.2. Cellular and Animal TARDBP Model of ALS
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Compound Preparation
4.3. Mitophagy Assay
4.4. Immunostaining
4.5. Image Acquisition
4.6. Western Blotting
4.7. Biochemical Kinase Assay
4.8. Animal Samples Analysis
4.9. Histological Procedures
4.10. Image Acquisition and Analysis
4.11. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharm. 2018, 104, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madruga, E.; Maestro, I.; Martinez, A. Mitophagy Modulation, a New Player in the Race against ALS. Int. J. Mol. Sci. 2021, 22, 740. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Nagy, M.; Fenton, W.A.; Horwich, A.L. Absence of lipofuscin in motor neurons of SOD1-linked ALS mice. Proc. Natl. Acad. Sci. USA 2014, 111, 11055–11060. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.K.H.; Thombre, R.; Wang, J. Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 697, 34–48. [Google Scholar] [CrossRef]
- Xie, Y.; Zhou, B.; Lin, M.Y.; Wang, S.; Foust, K.D.; Sheng, Z.H. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron 2015, 87, 355–370. [Google Scholar] [CrossRef] [Green Version]
- Vicencio, E.; Beltran, S.; Labrador, L.; Manque, P.; Nassif, M.; Woehlbier, U. Implications of Selective Autophagy Dysfunction for ALS Pathology. Cells 2020, 9, 381. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; Porras, G.; Arribas-Blazquez, M.; Maestro, I.; Borrego-Hernandez, D.; Boya, P.; Cerdan, S.; Garcia-Redondo, A.; Martinez, A.; Martin-Requero, A. Molecular Alterations in Sporadic and SOD1-ALS Immortalized Lymphocytes: Towards a Personalized Therapy. Int. J. Mol. Sci. 2021, 22, 3007. [Google Scholar] [CrossRef]
- Sebastian-Perez, V.; Roca, C.; Awale, M.; Reymond, J.L.; Martinez, A.; Gil, C.; Campillo, N.E. Medicinal and Biological Chemistry (MBC) Library: An Efficient Source of New Hits. J. Chem. Inf. Model 2017, 57, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Princely Abudu, Y.; Pankiv, S.; Mathai, B.J.; Hakon Lystad, A.; Bindesboll, C.; Brenne, H.B.; Yoke Wui Ng, M.; Thiede, B.; Yamamoto, A.; Mutugi Nthiga, T.; et al. NIPSNAP1 and NIPSNAP2 Act as “Eat Me” Signals for Mitophagy. Dev. Cell 2019, 49, 509–525.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, G.F.; Toth, R.; James, J.; Ganley, I.G. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Munson, M.J.; Mathai, B.J.; Ng, M.Y.W.; Trachsel-Moncho, L.; de la Ballina, L.R.; Schultz, S.W.; Aman, Y.; Lystad, A.H.; Singh, S.; Singh, S.; et al. GAK and PRKCD are positive regulators of PRKN-independent mitophagy. Nat. Commun. 2021, 12, 6101. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Hoffbrand, A.V. The Role of Deferiprone in Iron Chelation. N. Engl. J. Med. 2018, 379, 2140–2150. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Yoshimori, T.; Yamamoto, A.; Moriyama, Y.; Futai, M.; Tashiro, Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991, 266, 17707–17712. [Google Scholar] [CrossRef]
- Salado, I.G.; Redondo, M.; Bello, M.L.; Perez, C.; Liachko, N.F.; Kraemer, B.C.; Miguel, L.; Lecourtois, M.; Gil, C.; Martinez, A.; et al. Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. J. Med. Chem. 2014, 57, 2755–2772. [Google Scholar] [CrossRef]
- Kametani, F.; Nonaka, T.; Suzuki, T.; Arai, T.; Dohmae, N.; Akiyama, H.; Hasegawa, M. Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem. Biophys. Res. Commun. 2009, 382, 405–409. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar]
- Martinez-Gonzalez, L.; Rodriguez-Cueto, C.; Cabezudo, D.; Bartolome, F.; Andres-Benito, P.; Ferrer, I.; Gil, C.; Martin-Requero, A.; Fernandez-Ruiz, J.; Martinez, A.; et al. Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1delta kinase inhibitor treatment. Sci. Rep. 2020, 10, 4449. [Google Scholar] [CrossRef] [PubMed]
- Posa, D.; Martinez-Gonzalez, L.; Bartolome, F.; Nagaraj, S.; Porras, G.; Martinez, A.; Martin-Requero, A. Recapitulation of pathological TDP-43 features in immortalized lymphocytes from sporadic ALS patients. Mol. Neurobiol. 2019, 56, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alquezar, C.; Salado, I.G.; de la Encarnacion, A.; Perez, D.I.; Moreno, F.; Gil, C.; de Munain, A.L.; Martinez, A.; Martin-Requero, A. Targeting TDP-43 phosphorylation by Casein Kinase-1delta inhibitors: A novel strategy for the treatment of frontotemporal dementia. Mol. Neurodegener. 2016, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Kocak, M.; Ezazi Erdi, S.; Jorba, G.; Maestro, I.; Farres, J.; Kirkin, V.; Martinez, A.; Pless, O. Targeting autophagy in disease: Established and new strategies. Autophagy 2022, 18, 473–495. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Wang, X.; Yu, X.; Duan, W.; Hong, K.; Wang, J.; Han, H.; Li, C. Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience 2015, 298, 12–25. [Google Scholar] [CrossRef]
- Zhou, Q.M.; Zhang, J.J.; Li, S.; Chen, S.; Le, W.D. n-butylidenephthalide treatment prolongs life span and attenuates motor neuron loss in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. CNS Neurosci. Ther. 2017, 23, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Maestro, I.; de la Ballina, L.R.; Simonsen, A.; Boya, P.; Martinez, A. Phenotypic Assay Leads to Discovery of Mitophagy Inducers with Therapeutic Potential for Parkinson’s Disease. ACS Chem. Neurosci. 2021, 12, 4512–4523. [Google Scholar] [CrossRef]
- Palomo, V.; Nozal, V.; Rojas-Prats, E.; Gil, C.; Martinez, A. Protein kinase inhibitors for amyotrophic lateral sclerosis therapy. Br. J. Pharmacol. 2021, 178, 1316–1335. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, X.; Xiong, Q.; Chen, Y.; Zhao, H.; Tahir, M.; Song, J.; Zhou, B.; Wang, J. Casein Kinase 1 Family Member CK1delta/Hrr25 Is Required for Autophagosome Completion. Front. Cell Dev. Biol. 2020, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int J Mol Sci 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Martinez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Marino, G.; Seco, E.; Durand, S.; Enot, D.; Grana, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef] [PubMed]
- Domenech, E.; Maestre, C.; Esteban-Martinez, L.; Partida, D.; Pascual, R.; Fernandez-Miranda, G.; Seco, E.; Campos-Olivas, R.; Perez, M.; Megias, D.; et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell Biol. 2015, 17, 1304–1316. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.proteinatlas.org/ENSG00000185345-PRKN/tissue (accessed on 4 March 2022).
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Agosta, F.; Al-Chalabi, A.; Filippi, M.; Hardiman, O.; Kaji, R.; Meininger, V.; Nakano, I.; Shaw, P.; Shefner, J.; van den Berg, L.H.; et al. The El Escorial criteria: Strengths and weaknesses. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 1–7. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maestro, I.; de la Ballina, L.R.; Porras, G.; Corrochano, S.; De Lago, E.; Simonsen, A.; Boya, P.; Martinez, A. Discovery of Mitophagy Inhibitors with Therapeutic Potential in Different Familial Amyotrophic Lateral Sclerosis Mutations. Int. J. Mol. Sci. 2022, 23, 12676. https://doi.org/10.3390/ijms232012676
Maestro I, de la Ballina LR, Porras G, Corrochano S, De Lago E, Simonsen A, Boya P, Martinez A. Discovery of Mitophagy Inhibitors with Therapeutic Potential in Different Familial Amyotrophic Lateral Sclerosis Mutations. International Journal of Molecular Sciences. 2022; 23(20):12676. https://doi.org/10.3390/ijms232012676
Chicago/Turabian StyleMaestro, Ines, Laura R. de la Ballina, Gracia Porras, Silvia Corrochano, Eva De Lago, Anne Simonsen, Patricia Boya, and Ana Martinez. 2022. "Discovery of Mitophagy Inhibitors with Therapeutic Potential in Different Familial Amyotrophic Lateral Sclerosis Mutations" International Journal of Molecular Sciences 23, no. 20: 12676. https://doi.org/10.3390/ijms232012676
APA StyleMaestro, I., de la Ballina, L. R., Porras, G., Corrochano, S., De Lago, E., Simonsen, A., Boya, P., & Martinez, A. (2022). Discovery of Mitophagy Inhibitors with Therapeutic Potential in Different Familial Amyotrophic Lateral Sclerosis Mutations. International Journal of Molecular Sciences, 23(20), 12676. https://doi.org/10.3390/ijms232012676