
Bacteriophage-Mediated Cancer Gene Therapy


Abstract
:1. Introduction
2. Bacteriophages: Structure and Biology
3. Bacteriophage–Eukaryotic Cells Interactions
3.1. Binding and Entry of Phage into Eukaryotic Cells
3.2. Phage Intracellular Activity
4. Bacteriophage-Mediated Cancer Gene Therapy
4.1. Phage Display
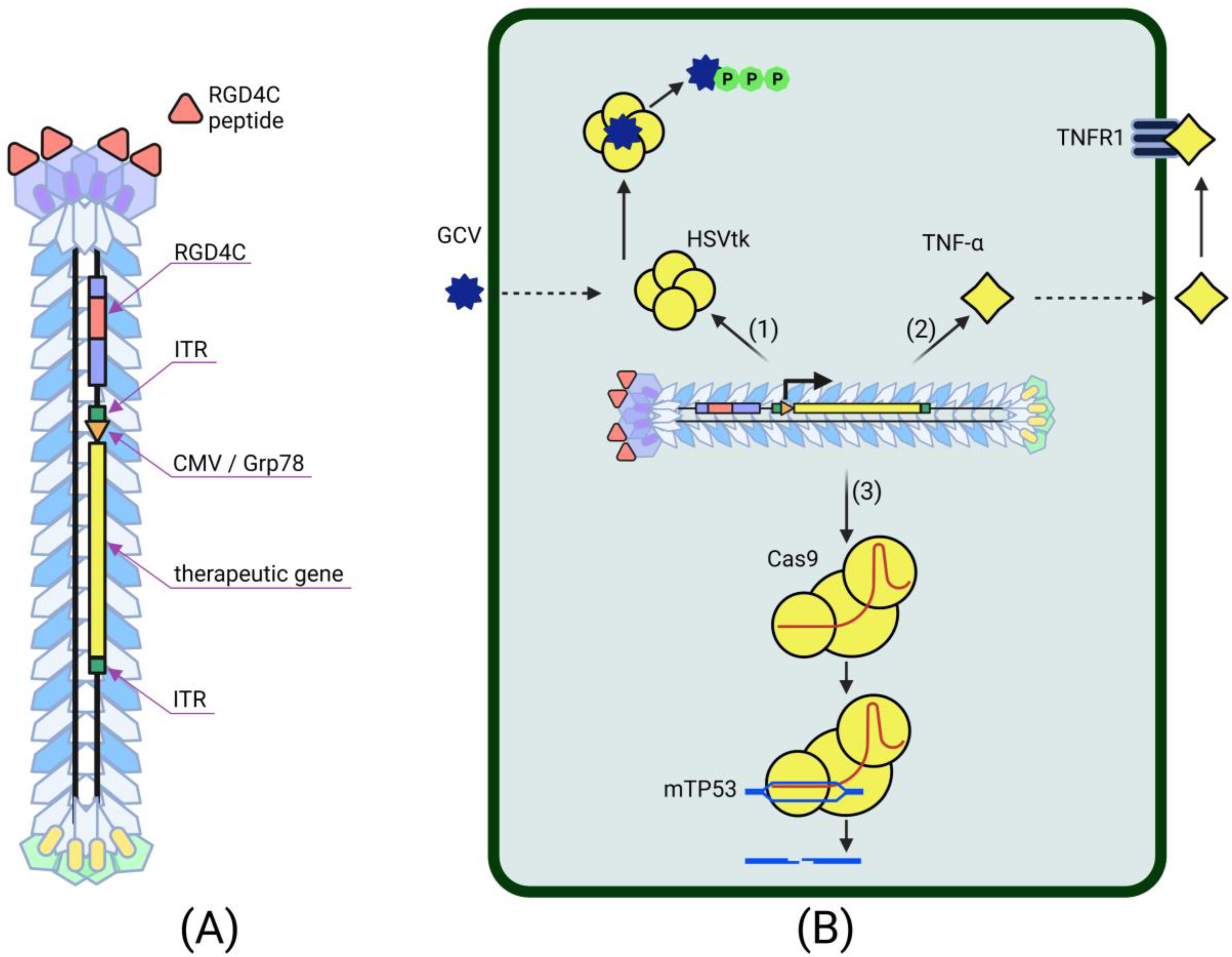
4.2. Phage-Based Vectors for Cancer Gene Therapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Curiel, D.T. Cancer gene therapy. Technol. Cancer Res. Treat. 2005, 4, 315–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouard, D.; Alazard-Dany, D.; Cosset, F.L. Viral vectors: From virology to transgene expression. Br. J. Pharmacol. 2009, 157, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waehler, R.; Russell, S.J.; Curiel, D.T. Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 2007, 8, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Hosseinidoust, Z. Phage-Mediated Gene Therapy. Curr. Gene Ther. 2017, 17, 120–126. [Google Scholar] [CrossRef]
- Hajitou, A. Targeted systemic gene therapy and molecular imaging of cancer contribution of the vascular-targeted AAVP vector. Adv. Genet. 2010, 69, 65–82. [Google Scholar] [CrossRef]
- Bao, Q.; Li, X.; Han, G.; Zhu, Y.; Mao, C.; Yang, M. Phage-based vaccines. Adv. Drug Deliv. Rev. 2019, 145, 40–56. [Google Scholar] [CrossRef]
- Zhang, J.; Li, D.; Zhang, R.; Peng, R.; Li, J. Delivery of microRNA-21-sponge and pre-microRNA-122 by MS2 virus-like particles to therapeutically target hepatocellular carcinoma cells. Exp. Biol. Med. 2021, 246, 2463–2472. [Google Scholar] [CrossRef]
- Chang, L.; Wang, G.; Jia, T.; Zhang, L.; Li, Y.; Han, Y.; Zhang, K.; Lin, G.; Zhang, R.; Li, J.; et al. Armored long non-coding RNA MEG3 targeting EGFR based on recombinant MS2 bacteriophage virus-like particles against hepatocellular carcinoma. Oncotarget 2016, 7, 23988–24004. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, L.; Tapia-Moreno, A.; Juarez-Moreno, K.; Patterson, D.P.; Cadena-Nava, R.D.; Douglas, T.; Vazquez-Duhalt, R. Design of a VLP-nanovehicle for CYP450 enzymatic activity delivery. J. Nanobiotechnol. 2015, 13, 66. [Google Scholar] [CrossRef]
- Gandra, N.; Abbineni, G.; Qu, X.; Huai, Y.; Wang, L.; Mao, C. Bacteriophage bionanowire as a carrier for both cancer-targeting peptides and photosensitizers and its use in selective cancer cell killing by photodynamic therapy. Small 2013, 9, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalao, M.J.; Gil, F.; Moniz-Pereira, J.; Sao-Jose, C.; Pimentel, M. Diversity in bacterial lysis systems: Bacteriophages show the way. FEMS Microbiol. Rev. 2013, 37, 554–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskella, B.; Brockhurst, M.A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef] [Green Version]
- Gibb, B.; Hyman, P.; Schneider, C.L. The Many Applications of Engineered Bacteriophages-An Overview. Pharmaceuticals 2021, 14, 634. [Google Scholar] [CrossRef] [PubMed]
- Lanigan, T.M.; Kopera, H.C.; Saunders, T.L. Principles of Genetic Engineering. Genes 2020, 11, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, P. The Bacteriophage Head-to-Tail Interface. Subcell. Biochem. 2018, 88, 305–328. [Google Scholar] [CrossRef]
- Kostyuchenko, V.A.; Leiman, P.G.; Chipman, P.R.; Kanamaru, S.; van Raaij, M.J.; Arisaka, F.; Mesyanzhinov, V.V.; Rossmann, M.G. Three-dimensional structure of bacteriophage T4 baseplate. Nat. Struct. Mol. Biol. 2003, 10, 688–693. [Google Scholar] [CrossRef]
- Boeckaerts, D.; Stock, M.; Criel, B.; Gerstmans, H.; De Baets, B.; Briers, Y. Predicting bacteriophage hosts based on sequences of annotated receptor-binding proteins. Sci. Rep. 2021, 11, 1467. [Google Scholar] [CrossRef]
- Straus, S.K.; Bo, H.E. Filamentous Bacteriophage Proteins and Assembly. Subcell. Biochem. 2018, 88, 261–279. [Google Scholar] [CrossRef]
- Stone, E.; Campbell, K.; Grant, I.; McAuliffe, O. Understanding and Exploiting Phage-Host Interactions. Viruses 2019, 11, 567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiman, P.G.; Shneider, M.M. Contractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 2012, 726, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Dams, D.; Brondsted, L.; Drulis-Kawa, Z.; Briers, Y. Engineering of receptor-binding proteins in bacteriophages and phage tail-like bacteriocins. Biochem. Soc. Trans. 2019, 47, 449–460. [Google Scholar] [CrossRef]
- Sciara, G.; Bebeacua, C.; Bron, P.; Tremblay, D.; Ortiz-Lombardia, M.; Lichière, J.; van Heel, M.; Campanacci, V.; Moineau, S.; Cambillau, C. Structure of lactococcal phage p2 baseplate and its mechanism of activation. Proc. Natl. Acad. Sci. USA 2010, 107, 6852–6857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szermer-Olearnik, B.; Drab, M.; Mąkosa, M.; Zembala, M.; Barbasz, J.; Dąbrowska, K.; Boratyński, J. Aggregation/dispersion transitions of T4 phage triggered by environmental ion availability. J. Nanobiotechnol. 2017, 15, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maghsoodi, A.; Chatterjee, A.; Andricioaei, I.; Perkins, N.C. How the phage T4 injection machinery works including energetics, forces, and dynamic pathway. Proc. Natl. Acad. Sci. USA 2019, 116, 25097–25105. [Google Scholar] [CrossRef] [PubMed]
- Hay, I.D.; Lithgow, T. Filamentous phages: Masters of a microbial sharing economy. EMBO Rep. 2019, 20, e47427. [Google Scholar] [CrossRef] [PubMed]
- Olszak, T.; Latka, A.; Roszniowski, B.; Valvano, M.A.; Drulis-Kawa, Z. Phage Life Cycles Behind Bacterial Biodiversity. Curr. Med. Chem. 2017, 24, 3987–4001. [Google Scholar] [CrossRef] [Green Version]
- Lobocka, M.; Dabrowska, K.; Gorski, A. Engineered Bacteriophage Therapeutics: Rationale, Challenges and Future. BioDrugs 2021, 35, 255–280. [Google Scholar] [CrossRef]
- Lang, L.H. FDA approves use of bacteriophages to be added to meat and poultry products. Gastroenterology 2006, 131, 1370. [Google Scholar] [CrossRef]
- Svircev, A.; Roach, D.; Castle, A. Framing the Future with Bacteriophages in Agriculture. Viruses 2018, 10, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, D.A.; Sharp, N.J.; Westwater, C. Phage-based platforms for the clinical detection of human bacterial pathogens. Bacteriophage 2012, 2, 105–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, A. The future of bacteriophage biology. Nat. Rev. Genet. 2003, 4, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Podlacha, M.; Grabowski, Ł.; Kosznik-Kawśnicka, K.; Zdrojewska, K.; Stasiłojć, M.; Węgrzyn, G.; Węgrzyn, A. Interactions of Bacteriophages with Animal and Human Organisms-Safety Issues in the Light of Phage Therapy. Int. J. Mol. Sci. 2021, 22, 8937. [Google Scholar] [CrossRef] [PubMed]
- Bakhshinejad, B.; Sadeghizadeh, M. Bacteriophages as vehicles for gene delivery into mammalian cells: Prospects and problems. Expert Opin. Drug Deliv. 2014, 11, 1561–1574. [Google Scholar] [CrossRef]
- Kurzepa, A.; Dabrowska, K.; Skaradzinski, G.; Gorski, A. Bacteriophage interactions with phagocytes and their potential significance in experimental therapy. Clin. Exp. Med. 2009, 9, 93–100. [Google Scholar] [CrossRef]
- Merril, C.R.; Friedman, T.B.; Attallah, A.F.M.; Geier, M.R.; Krell, K.; Yarkin, R. Isolation of bacteriophages from commercial sera. In Vitr. Cell. Dev. Biol. 1972, 8, 91–93. [Google Scholar] [CrossRef]
- Geier, M.R.; Merril, C.R. Lambda phage transcription in human fibroblasts. Virology 1972, 47, 638–643. [Google Scholar] [CrossRef]
- Larocca, D.; Witte, A.; Johnson, W.; Pierce, G.F.; Baird, A. Targeting bacteriophage to mammalian cell surface receptors for gene delivery. Hum. Gene Ther. 1998, 9, 2393–2399. [Google Scholar] [CrossRef]
- Gorski, A.; Dabrowska, K.; Switala-Jeleń, K.; Nowaczyk, M.; Weber-Dabrowska, B.; Boratynski, J.; Wietrzyk, J.; Opolski, A. New insights into the possible role of bacteriophages in host defense and disease. Med. Immunol. 2003, 2, 2. [Google Scholar] [CrossRef]
- Dabrowska, K.; Opolski, A.; Wietrzyk, J.; Switala-Jelen, K.; Boratynski, J.; Nasulewicz, A.; Lipinska, L.; Chybicka, A.; Kujawa, M.; Zabel, M.; et al. Antitumor activity of bacteriophages in murine experimental cancer models caused possibly by inhibition of beta3 integrin signaling pathway. Acta Virol. 2004, 48, 241–248. [Google Scholar] [PubMed]
- Sanmukh, S.G.; Dos Santos SA, A.; Felisbino, S.L. Natural bacteriophages T4 and M13 down-regulates Hsp90 gene expression in human prostate cancer cells (PC-3) representing a potential nanoparticle against cancer. Virol. Res. J. 2017, 1, 21–23. [Google Scholar]
- Lehti, T.A.; Pajunen, M.I.; Skog, M.S.; Finne, J. Internalization of a polysialic acid-binding Escherichia coli bacteriophage into eukaryotic neuroblastoma cells. Nat. Commun. 2017, 8, 1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, S.; Baker, K.; Padman, B.S.; Patwa, R.; Dunstan, R.A.; Weston, T.A.; Schlosser, S.; Bailey, B.; Lithgow, T.; Lazarou, M.; et al. Bacteriophage Transcytosis Provides a Mechanism To Cross Epithelial Cell Layers. mBio 2017, 8, e01874-17. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Wu, M.; Liu, X.; Liu, Z.; Zhou, Q.; Niu, Z.; Huang, Y. Probing the endocytic pathways of the filamentous bacteriophage in live cells using ratiometric pH fluorescent indicator. Adv. Healthc. Mater. 2015, 4, 413–419. [Google Scholar] [CrossRef]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363. [Google Scholar] [CrossRef]
- Bichet, M.C.; Chin, W.H.; Richards, W.; Lin, Y.-W.; Avellaneda-Franco, L.; Hernandez, C.A.; Oddo, A.; Chernyavskiy, O.; Hilsenstein, V.; Neild, A.; et al. Bacteriophage uptake by mammalian cell layers represents a potential sink that may impact phage therapy. iScience 2021, 24, 102287. [Google Scholar] [CrossRef]
- Bodner, K.; Melkonian, A.L.; Covert, M.W. The Enemy of My Enemy: New Insights Regarding Bacteriophage-Mammalian Cell Interactions. Trends Microbiol. 2021, 29, 528–541. [Google Scholar] [CrossRef]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [Green Version]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Hulo, C.; Masson, P.; de Castro, E.; Auchincloss, A.H.; Foulger, R.; Poux, S.; Lomax, J.; Bougueleret, L.; Xenarios, I.; Le Mercier, P. The ins and outs of eukaryotic viruses: Knowledge base and ontology of a viral infection. PLoS ONE 2017, 12, e0171746. [Google Scholar] [CrossRef] [PubMed]
- Duerkop, B.A.; Hooper, L.V. Resident viruses and their interactions with the immune system. Nat. Immunol. 2013, 14, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Volcy, K.; Dewhurst, S. Proteasome inhibitors enhance bacteriophage lambda (lambda) mediated gene transfer in mammalian cells. Virology 2009, 384, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Przystal, J.M.; Umukoro, E.; Stoneham, C.A.; Yata, T.; O’Neill, K.; Syed, N.; Hajitou, A. Proteasome inhibition in cancer is associated with enhanced tumor targeting by the adeno-associated virus/phage. Mol. Oncol. 2013, 7, 55–66. [Google Scholar] [CrossRef]
- Tsafa, E.; Al-Bahrani, M.; Bentayebi, K.; Przystal, J.; Suwan, K.; Hajitou, A. The natural dietary genistein boosts bacteriophage-mediated cancer cell killing by improving phage-targeted tumor cell transduction. Oncotarget 2016, 7, 52135–52149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fay, N.; Pante, N. Nuclear entry of DNA viruses. Front. Microbiol. 2015, 6, 467. [Google Scholar] [CrossRef] [PubMed]
- Redrejo-Rodriguez, M.; Munoz-Espin, D.; Holguera, I.; Mencia, M.; Salas, M. Functional eukaryotic nuclear localization signals are widespread in terminal proteins of bacteriophages. Proc. Natl. Acad. Sci. USA 2012, 109, 18482–18487. [Google Scholar] [CrossRef] [Green Version]
- Redrejo-Rodriguez, M.; Salas, M. Multiple roles of genome-attached bacteriophage terminal proteins. Virology 2014, 468–470, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Merril, C.R.; Geier, M.R.; Petricciani, J.C. Bacterial virus gene expression in human cells. Nature 1971, 233, 398–400. [Google Scholar] [CrossRef]
- Bentancor, L.V.; Bilen, M.F.; Mejías, M.P.; Fernández-Brando, R.J.; Panek, C.A.; Ramos, M.V.; Fernandez, G.C.; Isturiz, M.; Ghiringhelli, P.D.; Palermo, M.S. Functional capacity of Shiga-toxin promoter sequences in eukaryotic cells. PLoS ONE 2013, 8, e57128. [Google Scholar] [CrossRef] [Green Version]
- Sanmukh, S.G.; dos Santos, N.J.; Barquilha, C.N.; Cucielo, M.S.; de Carvalho, M.; dos Reis, P.P.; Delella, F.K.; Carvalho, H.F.; Felisbino, S.L. Bacteriophages M13 and T4 Increase the Expression of Anchorage-Dependent Survival Pathway Genes and Down Regulate Androgen Receptor Expression in LNCaP Prostate Cell Line. Viruses 2021, 13, 1754. [Google Scholar] [CrossRef] [PubMed]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Haq, I.U.; Chaudhry, W.N.; Akhtar, M.N.; Andleeb, S.; Qadri, I. Bacteriophages and their implications on future biotechnology: A review. Virol. J. 2012, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv. 2010, 28, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Rakonjac, J.; Russel, M.; Khanum, S.; Brooke, S.J.; Rajic, M. Filamentous Phage: Structure and Biology. Adv. Exp. Med. Biol. 2017, 1053, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, V.A. Landscape Phage: Evolution from Phage Display to Nanobiotechnology. Viruses 2018, 10, 311. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Tian, T.; Liu, W.; Zhu, Z.; Yang, C.J. Advance in phage display technology for bioanalysis. Biotechnol. J. 2016, 11, 732–745. [Google Scholar] [CrossRef]
- Sakamoto, K.; Kamada, Y.; Sameshima, T.; Yaguchi, M.; Niida, A.; Sasaki, S.; Miwa, M.; Ohkubo, S.; Sakamoto, J.-I.; Kamaura, M.; et al. K-Ras(G12D)-selective inhibitory peptides generated by random peptide T7 phage display technology. Biochem. Biophys. Res. Commun. 2017, 484, 605–611. [Google Scholar] [CrossRef]
- Zuo, S.; Dai, G.; Wang, L.; Wen, Y.; Huang, Z.; Yang, W.; Ma, W.; Ren, X. Suppression of angiogenesis and tumor growth by recombinant T4 phages displaying extracellular domain of vascular endothelial growth factor receptor 2. Arch. Virol. 2019, 164, 69–82. [Google Scholar] [CrossRef]
- Aghebati-Maleki, L.; Bakhshinejad, B.; Baradaran, B.; Motallebnezhad, M.; Aghebati-Maleki, A.; Nickho, H.; Yousefi, M.; Majidi, J. Phage display as a promising approach for vaccine development. J. Biomed. Sci. 2016, 23, 66. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lamolinara, A.; Conti, L.; Giangrossi, M.; Cui, L.; Morelli, M.B.; Amantini, C.; Falconi, M.; Bartolacci, C.; Andreani, C.; et al. HER2-Displaying M13 Bacteriophages induce Therapeutic Immunity against Breast Cancer. Cancers 2022, 14, 4054. [Google Scholar] [CrossRef] [PubMed]
- Iwagami, Y.; Casulli, S.; Nagaoka, K.; Kim, M.; Carlson, R.I.; Ogawa, K.; Lebowitz, M.S.; Fuller, S.; Biswas, B.; Stewart, S.; et al. Lambda phage-based vaccine induces antitumor immunity in hepatocellular carcinoma. Heliyon 2017, 3, e00407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajitou, A.; Trepel, M.; Lilley, C.E.; Soghomonyan, S.; Alauddin, M.M.; Marini, F.C.; Restel, B.H.; Ozawa, M.G.; Moya, C.A.; Rangel, R.; et al. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell 2006, 125, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Trepel, M.; Stoneham, C.A.; Eleftherohorinou, H.; Mazarakis, N.D.; Pasqualini, R.; Arap, W.; Hajitou, A. A heterotypic bystander effect for tumor cell killing after adeno-associated virus/phage-mediated, vascular-targeted suicide gene transfer. Mol. Cancer Ther. 2009, 8, 2383–2391. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.L.; Yuan, Z.; Cardó-Vila, M.; Claros, C.S.; Adem, A.; Cui, M.-H.; Branch, C.A.; Gelovani, J.G.; Libutti, S.K.; Sidman, R.L.; et al. AAVP displaying octreotide for ligand-directed therapeutic transgene delivery in neuroendocrine tumors of the pancreas. Proc. Natl. Acad. Sci. USA 2016, 113, 2466–2471. [Google Scholar] [CrossRef] [Green Version]
- Tandle, A.; Hanna, E.; Lorang, D.; Hajitou, A.; Moya, C.A.; Pasqualini, R.; Arap, W.; Adem, A.; Starker, E.; Hewitt, S.; et al. Tumor vasculature-targeted delivery of tumor necrosis factor-alpha. Cancer 2009, 115, 128–139. [Google Scholar] [CrossRef]
- Chongchai, A.; Waramit, S.; Suwan, K.; Al-Bahrani, M.; Udomruk, S.; Phitak, T.; Kongtawelert, P.; Pothacharoen, P.; Hajitou, A. Bacteriophage-mediated therapy of chondrosarcoma by selective delivery of the tumor necrosis factor alpha (TNFalpha) gene. FASEB J. 2021, 35, e21487. [Google Scholar] [CrossRef]
- Yang Zhou, J.; Suwan, K.; Hajitou, A. Initial Steps for the Development of a Phage-Mediated Gene Replacement Therapy Using CRISPR-Cas9 Technology. J. Clin. Med. 2020, 9, 1498. [Google Scholar] [CrossRef] [PubMed]
- Asavarut, P.; Waramit, S.; Suwan, K.; Marais, G.J.K.; Chongchai, A.; Benjathummarak, S.; Al-Bahrani, M.; Vila-Gomez, P.; Williams, M.; Kongtawelert, P.; et al. Systemically targeted cancer immunotherapy and gene delivery using transmorphic particles. EMBO Mol. Med. 2022, 14, e15418. [Google Scholar] [CrossRef]
- Hwang, Y.J.; Myung, H. Engineered Bacteriophage T7 as a Potent Anticancer Agent in vivo. Front. Microbiol. 2020, 11, 491001. [Google Scholar] [CrossRef]
- Yue, H.; Li, Y.; Yang, M.; Mao, C. T7 Phage as an Emerging Nanobiomaterial with Genetically Tunable Target Specificity. Adv. Sci. 2022, 9, 2103645. [Google Scholar] [CrossRef]
- Kaufmann, K.B.; Buning, H.; Galy, A.; Schambach, A.; Grez, M. Gene therapy on the move. EMBO Mol. Med. 2013, 5, 1642–1661. [Google Scholar] [CrossRef]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy- an overview. J. Clin. Diagn. Res. 2015, 9, GE01–GE06. [Google Scholar] [CrossRef]
- Larocca, D.; Kassner, P.D.; Witte, A.; Ladner, R.C.; Pierce, G.F.; Baird, A. Gene transfer to mammalian cells using genetically targeted filamentous bacteriophage. FASEB J. 1999, 13, 727–734. [Google Scholar] [CrossRef]
- Larocca, D.; Jensen-Pergakes, K.; Burg, M.A.; Baird, A. Receptor-targeted gene delivery using multivalent phagemid particles. Mol. Ther. 2001, 3, 476–484. [Google Scholar] [CrossRef]
- Hood, J.D.; Bednarski, M.; Frausto, R.; Guccione, S.; Reisfeld, R.A.; Xiang, R.; Cheresh, D.A. Tumor regression by targeted gene delivery to the neovasculature. Science 2002, 296, 2404–2407. [Google Scholar] [CrossRef] [Green Version]
- Hajitou, A.; Lev, D.C.; Hannay, J.A.F.; Korchin, B.; Staquicini, F.I.; Soghomonyan, S.; Alauddin, M.M.; Benjamin, R.S.; Pollock, R.E.; Gelovani, J.G.; et al. A preclinical model for predicting drug response in soft-tissue sarcoma with targeted AAVP molecular imaging. Proc. Natl. Acad. Sci. USA 2008, 105, 4471–4476. [Google Scholar] [CrossRef] [Green Version]
- Kia, A.; Przystal, J.M.; Nianiaris, N.; Mazarakis, N.D.; Mintz, P.J.; Hajitou, A. Dual systemic tumor targeting with ligand-directed phage and Grp78 promoter induces tumor regression. Mol. Cancer Ther. 2012, 11, 2566–2577. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Chang, Y.-N.; Ryan, P.; Linscott, M.; McGarrity, G.J.; Chiang, Y.L. Effect of herpes simplex virus thymidine kinase expression levels on ganciclovir-mediated cytotoxicity and the “bystander effect”. Hum. Gene Ther. 1995, 6, 1467–1476. [Google Scholar] [CrossRef]
- Sadeghi, H.; Hitt, M.M. Transcriptionally targeted adenovirus vectors. Curr. Gene Ther. 2005, 5, 411–427. [Google Scholar] [CrossRef]
- Li, J.; Lee, A.S. Stress induction of GRP78/BiP and its role in cancer. Curr. Mol. Med. 2006, 6, 45–54. [Google Scholar] [CrossRef]
- Przystal, J.M.; Waramit, S.; Pranjol, Z.I.; Yan, W.; Chu, G.; Chongchai, A.; Samarth, G.; Olaciregui, N.G.; Tabatabai, G.; Carcaboso, A.M.; et al. Efficacy of systemic temozolomide-activated phage-targeted gene therapy in human glioblastoma. EMBO Mol. Med. 2019, 11, e8492. [Google Scholar] [CrossRef]
- Pfaffenbach, K.T.; Lee, A.S. The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 2011, 23, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Stoneham, C.A.; Hollinshead, M.; Hajitou, A. Clathrin-mediated endocytosis and subsequent endo-lysosomal trafficking of adeno-associated virus/phage. J. Biol. Chem. 2012, 287, 35849–35859. [Google Scholar] [CrossRef] [Green Version]
- Yata, T.; Lee, E.L.Q.; Suwan, K.; Syed, N.; Asavarut, P.; Hajitou, A. Modulation of extracellular matrix in cancer is associated with enhanced tumor cell targeting by bacteriophage vectors. Mol. Cancer 2015, 14, 110. [Google Scholar] [CrossRef] [Green Version]
- Yata, T.; Lee, K.-Y.; Dharakul, T.; Songsivilai, S.; Bismarck, A.; Mintz, P.J.; Hajitou, A. Hybrid Nanomaterial Complexes for Advanced Phage-guided Gene Delivery. Mol. Ther. Nucleic Acids 2014, 3, e185. [Google Scholar] [CrossRef]
- Suwan, K.; Yata, T.; Waramit, S.; Przystal, J.M.; Stoneham, C.A.; Bentayebi, K.; Asavarut, P.; Chongchai, A.; Pothachareon, P.; Lee, K.-Y.; et al. Next-generation of targeted AAVP vectors for systemic transgene delivery against cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 18571–18577. [Google Scholar] [CrossRef] [Green Version]
- Josephs, S.F.; Ichim, T.E.; Prince, S.M.; Kesari, S.; Marincola, F.M.; Escobedo, A.R.; Jafri, A. Unleashing endogenous TNF-alpha as a cancer immunotherapeutic. J. Transl. Med. 2018, 16, 242. [Google Scholar] [CrossRef] [Green Version]
- Paoloni, M.C.; Tandle, A.; Mazcko, C.; Hanna, E.; Kachala, S.; Leblanc, A.; Newman, S.; Vail, D.; Henry, C.; Thamm, D.; et al. Launching a novel preclinical infrastructure: Comparative oncology trials consortium directed therapeutic targeting of TNFalpha to cancer vasculature. PLoS ONE 2009, 4, e4972. [Google Scholar] [CrossRef]
- Staquicini, F.I.; Smith, T.L.; Tang, F.H.F.; Gelovani, J.G.; Giordano, R.J.; Libutti, S.K.; Sidman, R.L.; Cavenee, W.K.; Arap, W.; Pasqualini, R. Targeted AAVP-based therapy in a mouse model of human glioblastoma: A comparison of cytotoxic versus suicide gene delivery strategies. Cancer Gene Ther. 2020, 27, 301–310. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef]
- Zhu, J.; Tao, P.; Mahalingam, M.; Sha, J.; Kilgore, P.; Chopra, A.K.; Rao, V. A prokaryotic-eukaryotic hybrid viral vector for delivery of large cargos of genes and proteins into human cells. Sci. Adv. 2019, 5, eaax0064l. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Bao, X.; Wang, Y.; Xu, Y.; Deng, B.; Lu, Y.; Hou, J. Engineering T7 bacteriophage as a potential DNA vaccine targeting delivery vector. Virol. J. 2018, 15, 49. [Google Scholar] [CrossRef]
- Sun, W.; Shi, Q.; Zhang, H.; Yang, K.; Ke, Y.; Wang, Y.; Qiao, L. Advances in the techniques and methodologies of cancer gene therapy. Discov. Med. 2019, 27, 45–55. [Google Scholar]
- Santiago-Ortiz, J.L.; Schaffer, D.V. Adeno-associated virus (AAV) vectors in cancer gene therapy. J. Control. Release 2016, 240, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Sagona, A.P.; Grigonyte, A.M.; MacDonald, P.R.; Jaramillo, A. Genetically modified bacteriophages. Integr. Biol. 2016, 8, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Pranjol, M.Z.; Hajitou, A. Bacteriophage-derived vectors for targeted cancer gene therapy. Viruses 2015, 7, 268–284. [Google Scholar] [CrossRef]
- Lankes, H.; Zanghi, C.; Santos, K.; Capella, C.; Duke, C.; Dewhurst, S. In vivo gene delivery and expression by bacteriophage lambda vectors. J. Appl. Microbiol. 2007, 102, 1337–1349. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Phage-Based Vector | Delivered Transgene | Advantages | Limitations | References |
|---|---|---|---|---|
| AAVP | HSVtk TNF-α CRISPR/Cas9 | Easy modifiability and production of viral particles; Lack of non-targeted expression | Initially low cell transduction efficiency compared to eukaryotic virus-based vectors; Increased phage capsid length, as a result of the insertion of large transgenic cassettes, leads to limitations in packaging, cloning capacity and susceptibility to clearance by the reticuloendothelial system | [74,75] [76,77,78] [79] |
| TPA | IL-12, IL-15 TNF-α | Increased efficiency of target cell transduction due to reduced viral particle size; Extremely high yield of viral particles | A helper phage is required for the production of TPA particles | [80] |
| T7 | GM-CSF | Large packaging capacity | Poor study as a vector for systemic delivery | [81,82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrov, G.; Dymova, M.; Richter, V. Bacteriophage-Mediated Cancer Gene Therapy. Int. J. Mol. Sci. 2022, 23, 14245. https://doi.org/10.3390/ijms232214245
Petrov G, Dymova M, Richter V. Bacteriophage-Mediated Cancer Gene Therapy. International Journal of Molecular Sciences. 2022; 23(22):14245. https://doi.org/10.3390/ijms232214245
Chicago/Turabian StylePetrov, Gleb, Maya Dymova, and Vladimir Richter. 2022. "Bacteriophage-Mediated Cancer Gene Therapy" International Journal of Molecular Sciences 23, no. 22: 14245. https://doi.org/10.3390/ijms232214245
APA StylePetrov, G., Dymova, M., & Richter, V. (2022). Bacteriophage-Mediated Cancer Gene Therapy. International Journal of Molecular Sciences, 23(22), 14245. https://doi.org/10.3390/ijms232214245