
Activation Mechanism of RhoA Caused by Constitutively Activating Mutations G14V and Q63L


Abstract
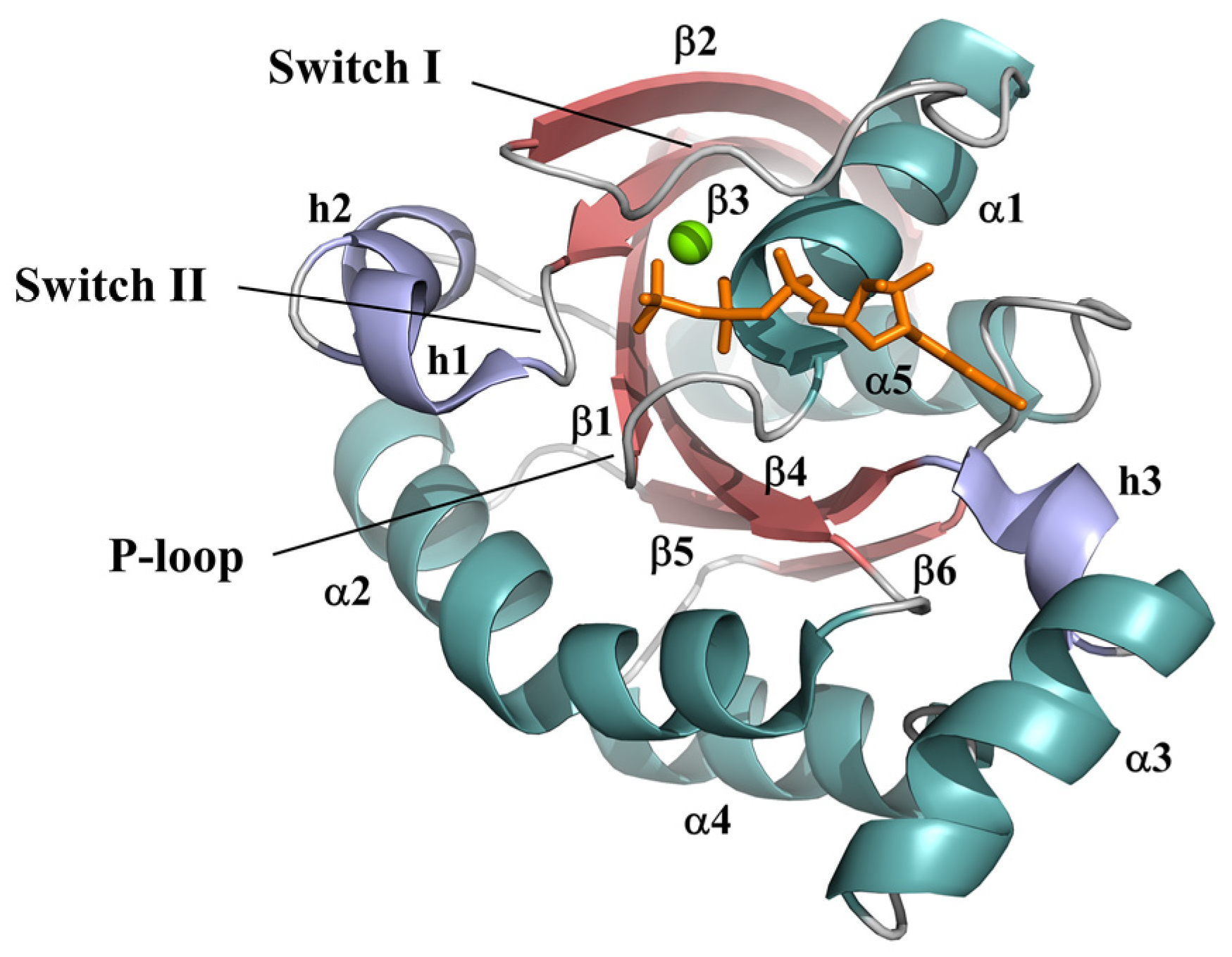
:1. Introduction
2. Results
2.1. Different Conformational Dynamics of the Switch Regions between the Wild-Type GTP-Bound RhoA and the Mutants
2.2. State Transition of the GTP-Bound RhoA Induced by Mutation
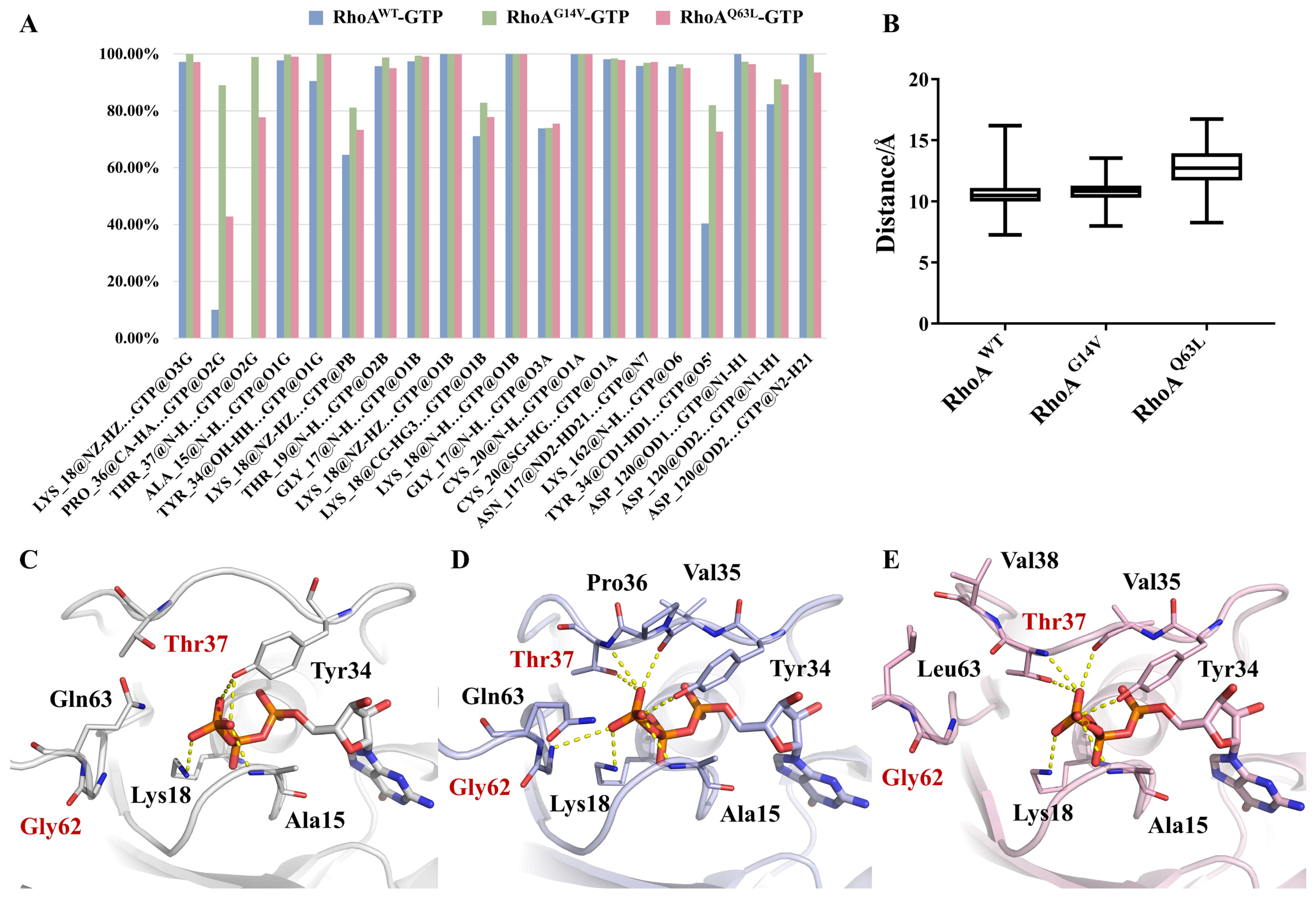
2.3. Significantly Increased Binding Affinity of GTP to Mutants G14V and Q63L
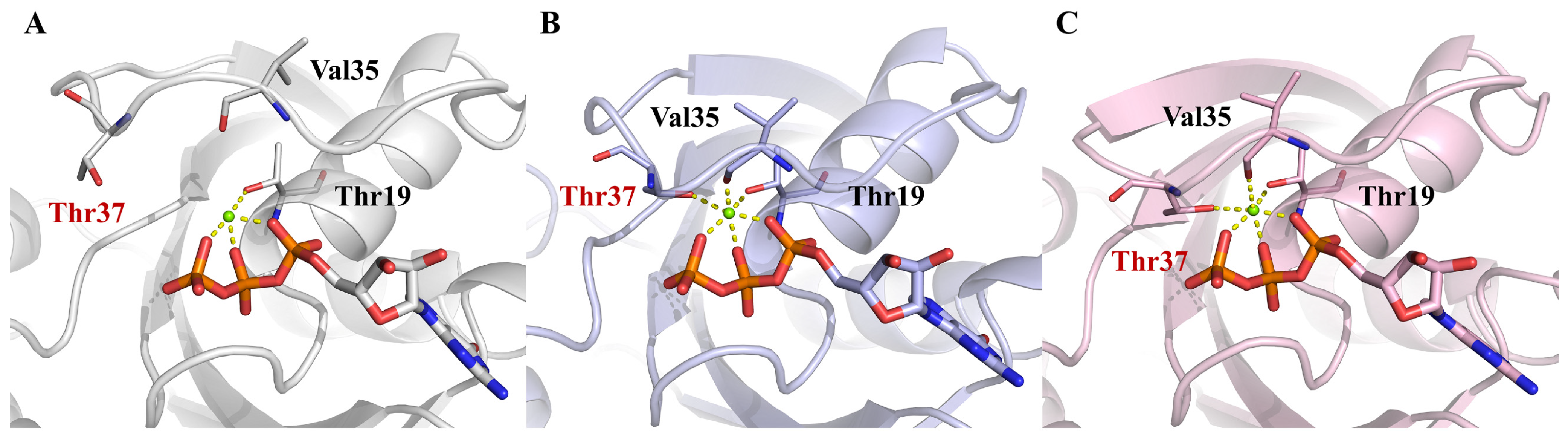
2.4. Interactions between RhoA and GTP around the Nucleotide-Binding Site
2.5. Changes in the Exposure of the GTP-Binding Site
3. Discussion
4. Materials and Methods
4.1. Preparation of Models
4.2. Simulation Protocol
4.3. MD Trajectory Analysis
4.3.1. RMSD/RMSF Calculation
4.3.2. SASA Calculation and Water Molecule Counting
4.3.3. Secondary Structure Assignment Analysis
4.3.4. Cluster Analysis
4.3.5. Interaction Analysis
4.4. Binding Free Energy Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sit, S.-T.; Manser, E. Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci. 2011, 124, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, S.; El-Sibai, M. Signaling networks of Rho GTPases in cell motility. Cell. Signal. 2013, 25, 1955–1961. [Google Scholar] [CrossRef]
- Mettouchi, A.; Klein, S.; Guo, W.J.; Lopez-Lago, M.; Lemichez, E.; Westwick, J.K.; Giancotti, F.G. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol. Cell 2001, 8, 115–127. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Hirshberg, M.; Stockley, R.W.; Dodson, G.; Webb, M.R. The crystal structure of human rac1, a member of the rho-family complexed with a GTP analogue. Nat. Struct. Biol. 1997, 4, 147–152. [Google Scholar] [CrossRef]
- Wei, Y.Y.; Zhang, Y.; Derewenda, U.; Liu, X.P.; Minor, W.; Nakamoto, R.K.; Somlyo, A.V.; Somlyo, A.P.; Derewenda, Z.S. Crystal structure of RhoA-GDP and its functional implications. Nat. Struct. Biol. 1997, 4, 699–703. [Google Scholar] [CrossRef]
- Ihara, K.; Muraguchi, S.; Kato, M.; Shimizu, T.; Shirakawa, M.; Kuroda, S.; Kaibuchi, K.; Hakoshima, T. Crystal structure of human RhoA in a dominantly active form complexed with a GTP analogue. J. Biol. Chem. 1998, 273, 9656–9666. [Google Scholar] [CrossRef] [Green Version]
- Longenecker, K.; Read, P.; Lin, S.K.; Somlyo, A.P.; Nakamoto, R.K.; Derewenda, Z.S. Structure of a constitutively activated RhoA mutant (Q63L) at 1.55 angstrom resolution. Acta Crystallogr. D 2003, 59, 876–880. [Google Scholar] [CrossRef]
- Cherfils, J.; Zeghouf, M. REGULATION OF SMALL GTPases BY GEFs, GAPs, AND GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [Green Version]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on Rho GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Olofsson, B. Rho guanine dissociation inhibitors: Pivotal molecules in cellular signalling. Cell. Signal. 1999, 11, 545–554. [Google Scholar] [CrossRef]
- Dovas, A.; Couchman, J.R. RhoGDI: Multiple functions in the regulation of Rho family GTPase activities. Biochem. J. 2005, 390, 1–9. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Milburn, M.V.; Tong, L.; de Vos, A.M.; Brunger, A.; Yamaizumi, Z.; Nishimura, S.; Kim, S.H. Molecular switch for signal transduction: Structural differences between active and inactive forms of protooncogenic ras proteins. Science 1990, 247, 939–945. [Google Scholar] [CrossRef] [Green Version]
- Pai, E.F.; Krengel, U.; Petsko, G.A.; Goody, R.S.; Kabsch, W.; Wittinghofer, A. Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: Implications for the mechanism of GTP hydrolysis. EMBO J. 1990, 9, 2351–2359. [Google Scholar] [CrossRef]
- Amor, J.C.; Harrison, D.H.; Kahn, R.A.; Ringe, D. Structure of the human ADP-ribosylation factor 1 complexed with GDP. Nature 1994, 372, 704–708. [Google Scholar] [CrossRef]
- Scheffzek, K.; Klebe, C.; Fritz-Wolf, K.; Kabsch, W.; Wittinghofer, A. Crystal structure of the nuclear Ras-related protein ran in its GDP-bound form. Nature 1995, 374, 378–381. [Google Scholar] [CrossRef]
- Nassar, N.; Horn, G.; Herrmann, C.; Scherer, A.; McCormick, F.; Wittinghofer, A. The 2.2 A crystal structure of the Ras-binding domain of serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature 1995, 375, 554–560. [Google Scholar] [CrossRef]
- Vetter, I.R.; Wittinghofer, A. Signal transduction—The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef]
- Kang, N.; Liu, J.; Zhao, Y. Dissociation mechanism of GDP from Cdc42 via DOCK9 revealed by molecular dynamics simulations. Proteins 2019, 87, 433–442. [Google Scholar] [CrossRef]
- Struckhoff, A.P.; Rana, M.K.; Worthylake, R.A. RhoA can lead the way in tumor cell invasion and metastasis. Front. Biosci. 2011, 16, 1915–1926. [Google Scholar] [CrossRef]
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 8, 733. [Google Scholar] [CrossRef] [Green Version]
- Vega, F.M.; Ridley, A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef] [Green Version]
- Ellenbroek, S.I.J.; Collard, J.G. Rho GTPases: Functions and association with cancer. Clin. Exp. Metastasis 2007, 24, 657–672. [Google Scholar] [CrossRef]
- Fritz, G.; Just, I.; Kaina, B. Rho GTPases are over-expressed in human tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef]
- Kamai, T.; Yamanishi, T.; Shirataki, H.; Takagi, K.; Asami, H.; Ito, Y.; Yoshida, K.I. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin. Cancer Res. 2004, 10, 4799–4805. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.T.; Kuriakose, M.A.; Sacks, P.G.; Yee, H.; Chiriboga, L.; Bearer, E.L.; Delacure, M.D. Motility-related proteins as markers for head and neck squamous cell cancer. Laryngoscope 2001, 111, 1285–1289. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef] [Green Version]
- Huff, L.P.; Decristo, M.J.; Trembath, D.; Kuan, P.F.; Yim, M.; Liu, J.; Cook, D.R.; Miller, C.R.; Der, C.J.; Cox, A.D. The Role of Ect2 Nuclear RhoGEF Activity in Ovarian Cancer Cell Transformation. Genes Cancer 2013, 4, 460–475. [Google Scholar] [CrossRef]
- Justilien, V.; Ali, S.A.; Jamieson, L.; Yin, N.; Cox, A.D.; Der, C.J.; Murray, N.R.; Fields, A.P. Ect2-Dependent rRNA Synthesis Is Required for KRAS-TRP53-Driven Lung Adenocarcinoma. Cancer Cell 2017, 31, 256–269. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Durkin, M.E.; Wang, D.; Tripathi, B.K.; Olson, L.; Yang, X.-Y.; Vass, W.C.; Popescu, N.C.; Lowy, D.R. Inactivation of the Dlc1 Gene Cooperates with Downregulation of p15(INK4b) and p16(Ink4a), Leading to Neoplastic Transformation and Poor Prognosis in Human Cancer. Cancer Res. 2012, 72, 5900–5911. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, G. A tumor suppressor DLC1: The functions and signal pathways. J. Cell. Physiol. 2020, 235, 4999–5007. [Google Scholar] [CrossRef]
- Mayer, T.; Meyer, M.; Janning, A.; Schiedel, A.C.; Barnekow, A. A mutant form of the rho protein can restore stress fibers and adhesion plaques in v-src transformed fibroblasts. Oncogene 1999, 18, 2117–2128. [Google Scholar] [CrossRef] [Green Version]
- Michaelson, D.; Silletti, J.; Murphy, G.; D’Eustachio, P.; Rush, M.; Philips, M.R. Differential localization of Rho GTPases in live cells: Regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 2001, 152, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Paterson, H.F.; Self, A.J.; Garrett, M.D.; Just, I.; Aktories, K.; Hall, A. Microinjection of recombinant p21rho induces rapid changes in cell morphology. J. Cell Biol. 1990, 111, 1001–1007. [Google Scholar] [CrossRef]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Kumawat, A.; Chakrabarty, S.; Kulkarni, K. Nucleotide Dependent Switching in Rho GTPase: Conformational Heterogeneity and Competing Molecular Interactions. Sci. Rep. 2017, 7, 45829. [Google Scholar] [CrossRef]
- Acuner, S.E.; Sumbul, F.; Torun, H.; Haliloglu, T. Oncogenic mutations on Rac1 affect global intrinsic dynamics underlying GTP and PAK1 binding. Biophys. J. 2021, 120, 866–876. [Google Scholar] [CrossRef]
- Haspel, N.; Jang, H.; Nussinov, R. Active and Inactive Cdc42 Differ in Their Insert Region Conformational Dynamics. Biophys. J. 2021, 120, 306–318. [Google Scholar] [CrossRef]
- Shima, F.; Ijiri, Y.; Muraoka, S.; Liao, J.; Ye, M.; Araki, M.; Matsumoto, K.; Yamamoto, N.; Sugimoto, T.; Yoshikawa, Y.; et al. Structural Basis for Conformational Dynamics of GTP-bound Ras Protein. J. Biol. Chem. 2010, 285, 22696–22705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, M.; Shima, F.; Yoshikawa, Y.; Muraoka, S.; Ijiri, Y.; Nagahara, Y.; Shirono, T.; Kataoka, T.; Tamura, A. Solution Structure of the State 1 Conformer of GTP-bound H-Ras Protein and Distinct Dynamic Properties between the State 1 and State 2 Conformers. J. Biol. Chem. 2011, 286, 39644–39653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Jang, H.; Muratcioglu, S.; Gursoy, A.; Keskin, O.; Nussinov, R.; Zhang, J. Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev. 2016, 116, 6607–6665. [Google Scholar] [CrossRef]
- Chen, S.; Shu, L.; Zhao, R.; Zhao, Y. Molecular dynamics simulations reveal the activation mechanism of mutations G12V and Q61L of Cdc42. Proteins 2022, 90, 1376–1389. [Google Scholar] [CrossRef]
- Liao, J.; Shima, F.; Araki, M.; Ye, M.; Muraoka, S.; Sugimoto, T.; Kawamura, M.; Yamamoto, N.; Tamura, A.; Kataoka, T. Two conformational states of Ras GTPase exhibit differential GTP-binding kinetics. Biochem. Biophys. Res. Commun. 2008, 369, 327–332. [Google Scholar] [CrossRef]
- Cavalli, A.; Carloni, P. Enzymatic GTP hydrolysis: Insights from an ab initio molecular dynamics study. J. Am. Chem. Soc. 2002, 124, 3763–3768. [Google Scholar] [CrossRef]
- Prakash, P.; Gorfe, A.A. Overview of simulation studies on the enzymatic activity and conformational dynamics of the GTPase Ras. Mol. Simul. 2014, 40, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Novelli, E.T.; First, J.T.; Webb, L.J. Quantitative Measurement of Intrinsic GTP Hydrolysis for Carcinogenic Glutamine 61 Mutants in H-Ras. Biochemistry 2018, 57, 6356–6366. [Google Scholar] [CrossRef]
- Li, R.; Zhang, B.L.; Zheng, Y. Structural determinants required for the interaction between rho GTPase and the GTPase-activating domain of p190. J. Biol. Chem. 1997, 272, 32830–32835. [Google Scholar] [CrossRef]
- Nusbaum, C.; Mikkelsen, T.S.; Zody, M.C.; Asakawa, S.; Taudien, S.; Garber, M.; Kodira, C.D.; Schueler, M.G.; Shimizu, A.; Whittaker, C.A.; et al. DNA sequence and analysis of human chromosome 8. Nature 2006, 439, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, I.; Hart, C.; Park, I.-H.; Shim, J.W. Genes causing congenital hydrocephalus: Their chromosomal characteristics of telomere proximity and DNA compositions. Exp. Neurol. 2021, 335, 113523. [Google Scholar] [CrossRef] [PubMed]
- Lucas, H.B.; McKnight, I.; Raines, R.; Hijazi, A.; Hart, C.; Lee, C.; Kim, D.-G.; Li, W.; Lee, P.H.U.; Shim, J.W. Factors Associated with Mutations: Their Matching Rates to Cardiovascular and Neurological Diseases. Int. J. Mol. Sci. 2021, 22, 5057. [Google Scholar] [CrossRef] [PubMed]
- Raines, R.; McKnight, I.; White, H.; Legg, K.; Lee, C.; Li, W.; Lee, P.H.U.; Shim, J.W. Drug-Targeted Genomes: Mutability of Ion Channels and GPCRs. Biomedicines 2022, 10, 594. [Google Scholar] [CrossRef]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Palomero, T.; Couronne, L.; Khiabanian, H.; Kim, M.-Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Yoo, H.Y.; Sung, M.K.; Lee, S.H.; Kim, S.; Lee, H.; Park, S.; Kim, S.C.; Lee, B.; Rho, K.; Lee, J.-E.; et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.H.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Wang, E.C.; Sun, H.Y.; Wang, J.M.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T.J. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate—DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Lemichez, E.; Aktories, K. Hijacking of Rho GTPases during bacterial infection. Exp. Cell Res. 2013, 319, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Azeez, K.R.A.; Knapp, S.; Fernandes, J.M.R.; Klussmann, E.; Elkins, J.M. The crystal structure of the RhoA-AKAP-Lbc DH-PH domain complex. Biochem. J. 2014, 464, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.P.; Karplus, M. Molecular switch in signal transduction: Reaction paths of the conformational changes in ras p21. Proc. Natl. Acad. Sci. USA 1997, 94, 11905–11910. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy Term | RhoAWT-GTP | RhoAG14V-GTP | RhoAQ63L-GTP |
|---|---|---|---|
| BFE Total | |||
| −227.08 ± 0.25 | −290.72 ± 0.55 | −262.51 ± 1.83 | |
| BFE per Residue | |||
| G14 (or V14) | −5.43 ± 0.20 | −13.46 ± 0.04 | −5.80 ± 0.18 |
| A15 | −7.19 ± 0.22 | −7.77 ± 0.17 | −7.66 ± 0.28 |
| C16 | −5.70 ± 0.07 | −6.16 ± 0.11 | −5.94 ± 0.31 |
| G17 | −7.82 ± 0.29 | −8.27 ± 0.20 | −8.14 ± 0.16 |
| K18 | −31.56 ± 0.54 | −42.73 ± 0.08 | −36.43 ± 0.65 |
| C20 | −7.95 ± 0.18 | −7.94 ± 0.06 | −7.95 ± 0.11 |
| Y34 | −11.14 ± 0.40 | −13.14 ± 0.06 | −12.06 ± 0.14 |
| Mg2+ | −30.59 ± 1.75 | −77.00 ± 0.88 | −67.04 ± 1.92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Zhang, Z.; Zhang, Y.; Choi, T.; Zhao, Y. Activation Mechanism of RhoA Caused by Constitutively Activating Mutations G14V and Q63L. Int. J. Mol. Sci. 2022, 23, 15458. https://doi.org/10.3390/ijms232415458
Chen S, Zhang Z, Zhang Y, Choi T, Zhao Y. Activation Mechanism of RhoA Caused by Constitutively Activating Mutations G14V and Q63L. International Journal of Molecular Sciences. 2022; 23(24):15458. https://doi.org/10.3390/ijms232415458
Chicago/Turabian StyleChen, Shiyao, Zirui Zhang, Yijing Zhang, Taeyoung Choi, and Yaxue Zhao. 2022. "Activation Mechanism of RhoA Caused by Constitutively Activating Mutations G14V and Q63L" International Journal of Molecular Sciences 23, no. 24: 15458. https://doi.org/10.3390/ijms232415458
APA StyleChen, S., Zhang, Z., Zhang, Y., Choi, T., & Zhao, Y. (2022). Activation Mechanism of RhoA Caused by Constitutively Activating Mutations G14V and Q63L. International Journal of Molecular Sciences, 23(24), 15458. https://doi.org/10.3390/ijms232415458