
Protein Dimerization via Tyr Residues: Highlight of a Slow Process with Co-Existence of Numerous Intermediates and Final Products

 , and
, and
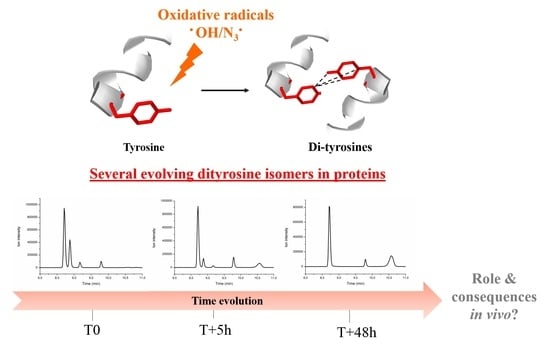
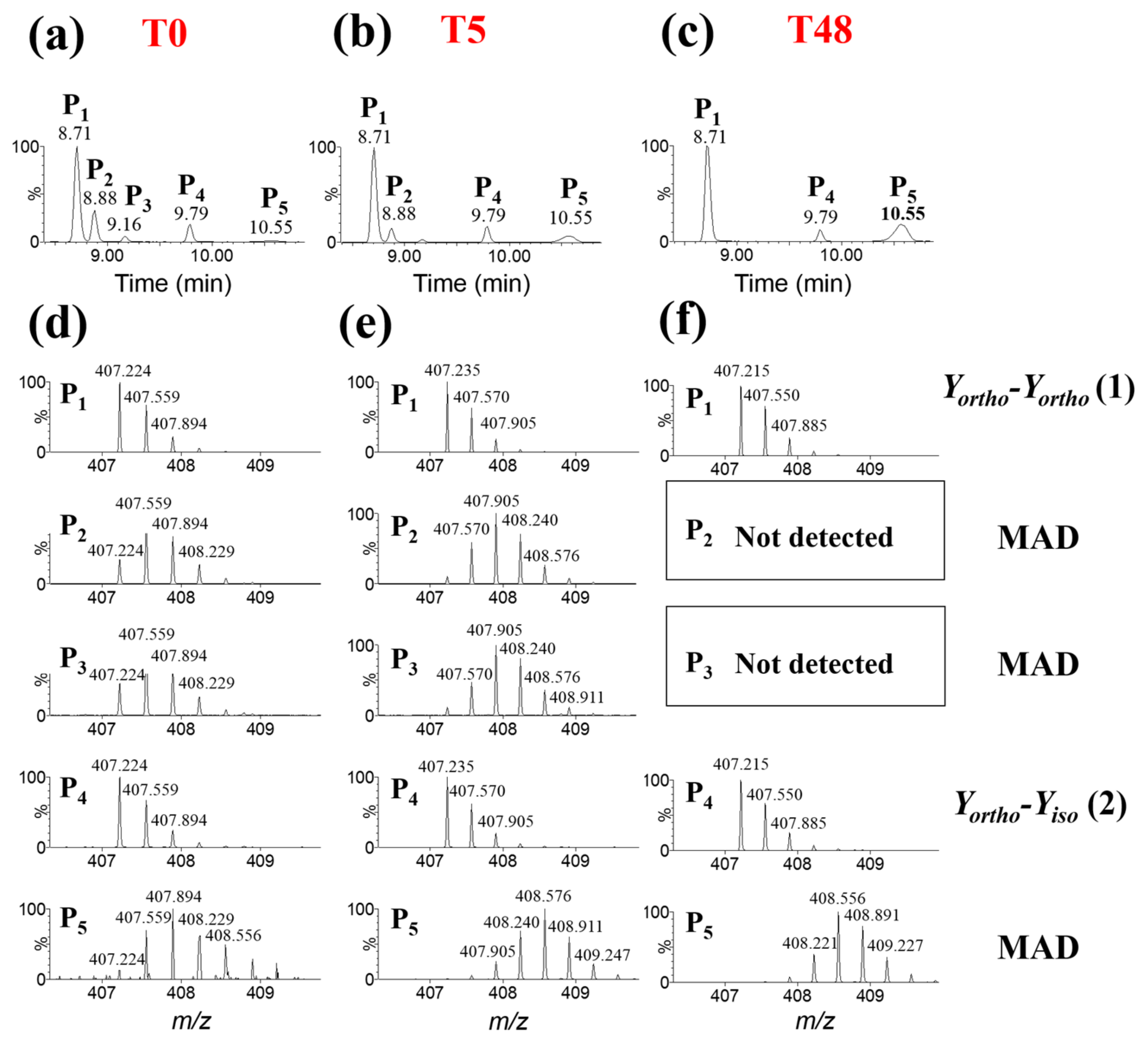
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Impact of the Amine Function Modification
2.1.1. N-Acetyl-L-Tyrosine (Ac-Y)
2.1.2. KTSLY Peptide
2.1.3. Human Centrin 2
2.2. Impact of the Carboxylic Function Modification
2.2.1. N-Acetyl-L-Tyrosine Ethyl Ester (Ac-Y-Et)
2.2.2. KTSLYG Peptide
2.2.3. Human Calmodulin
3. Discussion
4. Materials and Methods
4.1. Amino Acids and Peptides Preparation
4.2. Protein Production and Purification
4.2.1. Human Centrin 2
4.2.2. Human Calmodulin
4.3. Gamma Irradiation
4.4. Gel SDS-PAGE
4.5. Analysis: UPLC–MS
4.6. Data Treatment: Deuterium Average Incorporation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chatterjee, S. Oxidative Stress, Inflammation, and Disease. In Oxidative Stress and Biomaterials; Elsevier: Amsterdam, The Netherlands, 2016; pp. 35–58. ISBN 978-0-12-803269-5. [Google Scholar]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative Stress Pathways in Pancreatic β-Cells and Insulin-Sensitive Cells and Tissues: Importance to Cell Metabolism, Function, and Dysfunction. Am. J. Physiol. Cell Physiol. 2019, 317, C420–C433. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, P.; Rabbani, G.; Khan, R. The Role of Advanced Glycation End Products in Various Types of Neurodegenerative Disease: A Therapeutic Approach. Cell. Mol. Biol. Lett. 2014, 19, 407–437. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4+T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C.; Cunningham-Bussel, A. Beyond Oxidative Stress: An Immunologist’s Guide to Reactive Oxygen Species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, Oxidative Stress and Aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Detection, Identification, and Quantification of Oxidative Protein Modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef] [Green Version]
- Houée-Lévin, C.; Bobrowski, K.; Horakova, L.; Karademir, B.; Schöneich, C.; Davies, M.J.; Spickett, C.M. Exploring Oxidative Modifications of Tyrosine: An Update on Mechanisms of Formation, Advances in Analysis and Biological Consequences. Free Radic. Res. 2015, 49, 347–373. [Google Scholar] [CrossRef] [Green Version]
- Garrison, W.M. Reaction Mechanisms in the Radiolysis of Peptides, Polypeptides, and Proteins. Chem. Rev. 1987, 87, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Grune, T. Oxidized Protein Aggregates: Formation and Biological Effects. Free Radic. Biol. Med. 2020, 150, 120–124. [Google Scholar] [CrossRef]
- Davies, M.J.; Dean, R.T. Radical-Mediated Protein Oxidation: From Chemistry to Medicine; Oxford Science Publications; Oxford University Press: Oxford, NY, USA, 1997; ISBN 978-0-19-850097-1. [Google Scholar]
- Chen, Z.; Leinisch, F.; Greco, I.; Zhang, W.; Shu, N.; Chuang, C.Y.; Lund, M.N.; Davies, M.J. Characterisation and Quantification of Protein Oxidative Modifications and Amino Acid Racemisation in Powdered Infant Milk Formula. Free Radic. Res. 2019, 53, 68–81. [Google Scholar] [CrossRef]
- Fuentes-Lemus, E.; Silva, E.; Barrias, P.; Aspee, A.; Escobar, E.; Lorentzen, L.G.; Carroll, L.; Leinisch, F.; Davies, M.J.; López-Alarcón, C. Aggregation of α- and β- Caseins Induced by Peroxyl Radicals Involves Secondary Reactions of Carbonyl Compounds as Well as Di-Tyrosine and Di-Tryptophan Formation. Free Radic. Biol. Med. 2018, 124, 176–188. [Google Scholar] [CrossRef]
- Gatin, A.; Billault, I.; Duchambon, P.; Van der Rest, G.; Sicard-Roselli, C. Oxidative Radicals (HO• or N3•) Induce Several Di-Tyrosine Bridge Isomers at the Protein Scale. Free Radic. Biol. Med. 2021, 162, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Billault, I.; Gatin, A.; Van der Rest, G.; Sicard-Roselli, C. Advanced Methodology Combining UPLC-MS, Isotopic Labelling and H/D Exchanges Reveals Three Tyrosine-Tyrosine Cross-Links Induced by Oxidative Radicals Evolving to at Least Four Dimeric Structures. Anal. Bioanal. Chem. 2022, 414, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Solar, S.; Solar, W.; Getoff, N. Reactivity of Hydroxyl with Tyrosine in Aqueous Solution Studied by Pulse Radiolysis. J. Phys. Chem. 1984, 88, 2091–2095. [Google Scholar] [CrossRef]
- Land, E.J.; Prütz, W.A. Reaction of Azide Radicals with Amino Acids and Proteins. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1979, 36, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.D.; Sadler, I.H.; Fry, S.C. Di-Isodityrosine, a Novel Tetrametric Derivative of Tyrosine in Plant Cell Wall Proteins: A New Potential Cross-Link. Biochem. J. 1996, 315, 323–327. [Google Scholar] [CrossRef] [Green Version]
- Karam, L.R.; Dizdaroglu, M.; Simic, M.G. OH Radical-Induced Products of Tyrosine Peptides. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1984, 46, 715–724. [Google Scholar] [CrossRef]
- Fry, S.C. Isodityrosine, a New Cross-Linking Amino Acid from Plant Cell-Wall Glycoprotein. Biochem. J. 1982, 204, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Blouquit, Y.; Duchambon, P.; Brun, E.; Marco, S.; Rusconi, F.; Sicard-Roselli, C. High Sensitivity of Human Centrin 2 toward Radiolytical Oxidation: C-Terminal Tyrosinyl Residue as the Main Target. Free Radic. Biol. Med. 2007, 43, 216–228. [Google Scholar] [CrossRef]
- Brun, E.; Blouquit, Y.; Duchambon, P.; Malosse, C.; Chamot-Rooke, J.; Sicard-Roselli, C. Oxidative Stress Induces Mainly Human Centrin 2 Polymerisation. Int. J. Radiat. Biol. 2010, 86, 657–668. [Google Scholar] [CrossRef]
- Baxendale, I.R.; Ley, S.V.; Nessi, M.; Piutti, C. Total Synthesis of the Amaryllidaceae Alkaloid (+)-Plicamine Using Solid-Supported Reagents. Tetrahedron 2002, 58, 6285–6304. [Google Scholar] [CrossRef]
- Reddy, D.N.; Thirupathi, R.; Tumminakatti, S.; Prabhakaran, E.N. A Method for Stabilizing the Cis Prolyl Peptide Bond: Influence of an Unusual N→π∗ Interaction in 1,3-Oxazine and 1,3-Thiazine Containing Peptidomimetics. Tetrahedron Lett. 2012, 53, 4413–4417. [Google Scholar] [CrossRef]
- Babu, Y.S.; Bugg, C.E.; Cook, W.J. Structure of Calmodulin Refined at 2.2 Å Resolution. J. Mol. Biol. 1988, 204, 191–204. [Google Scholar] [CrossRef]
- Chattopadhyaya, R.; Meador, W.E.; Means, A.R.; Quiocho, F.A. Calmodulin Structure Refined at 1.7 Å Resolution. J. Mol. Biol. 1992, 228, 1177–1192. [Google Scholar] [CrossRef]
- Goldenberg, D.P.; Creighton, T.E. Gel Electrophoresis in Studies of Protein Conformation and Folding. Anal. Biochem. 1984, 138, 1–18. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Parsons-Mair, H.N.; Gebicki, S.; Gebicki, J.M.; Davies, M.J. Requirements for Superoxide-Dependent Tyrosine Hydroperoxide Formation in Peptides. Biochem. J. 2004, 381, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Resch, V.; Seidler, C.; Chen, B.-S.; Degeling, I.; Hanefeld, U. On the Michael Addition of Water to α,β-Unsaturated Ketones Using Amino Acids: Michael Addition of Water to α,β-Unsaturated Ketones. Eur. J. Org. Chem. 2013, 2013, 7697–7704. [Google Scholar] [CrossRef] [Green Version]
- Shamovsky, I.L.; Riopelle, R.J.; Ross, G.M. Ab Initio Studies on the Mechanism of Tyrosine Coupling. J. Phys. Chem. A 2001, 105, 1061–1070. [Google Scholar] [CrossRef]
- Malencik, D.A.; Anderson, S.R. Dityrosine Formation in Calmodulin: Conditions for Intermolecular Crosslinking. Biochemistry 1994, 33, 13363–13372. [Google Scholar] [CrossRef]
- Dahl, J.-U.; Gray, M.J.; Jakob, U. Protein Quality Control under Oxidative Stress Conditions. J. Mol. Biol. 2015, 427, 1549–1563. [Google Scholar] [CrossRef] [Green Version]
- Bobrowski, K.; Wierzchowski, K.L.; Holcman, J.; Ciurak, M. Intramolecular Electron Transfer in Peptides Containing Methionine, Tryptophan and Tyrosine: A Pulse Radiolysis Study. Int. J. Radiat. Biol. 1990, 57, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Prütz, W.A.; Land, E.J. Charge Transfer in Peptides: Pulse Radiolysis Investigation of One-Electron Reactions in Dipeptides of Tryptophan and Tyrosine. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1979, 36, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Land, E.J.; Prütz, W.A.; Swallow, A.J. Charge Transfer between Tryptophan and Tyrosine in Proteins. Biochim. Biophys. Acta BBA—Protein Struct. Mol. Enzymol. 1982, 705, 150–162. [Google Scholar] [CrossRef]
- Al-Hilaly, Y.K.; Williams, T.L.; Stewart-Parker, M.; Ford, L.; Skaria, E.; Cole, M.; Bucher, W.G.; Morris, K.L.; Sada, A.A.; Thorpe, J.R.; et al. A Central Role for Dityrosine Crosslinking of Amyloid-β in Alzheimer’s Disease. Acta Neuropathol. Commun. 2013, 1, 83. [Google Scholar] [CrossRef] [Green Version]
- Leeuwenburgh, C.; Rasmussen, J.E.; Hsu, F.F.; Mueller, D.M.; Pennathur, S.; Heinecke, J.W. Mass Spectrometric Quantification of Markers for Protein Oxidation by Tyrosyl Radical, Copper, and Hydroxyl Radical in Low Density Lipoprotein Isolated from Human Atherosclerotic Plaques. J. Biol. Chem. 1997, 272, 3520–3526. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Dozaki, N.; Nakamura, T.; Kitamoto, N.; Yoshida, A.; Naito, M.; Kitamura, M.; Osawa, T. Quantification of Modified Tyrosines in Healthy and Diabetic Human Urine Using Liquid Chromatography/Tandem Mass Spectrometry. J. Clin. Biochem. Nutr. 2009, 44, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Gibrat, G.; Assairi, F.L.; Blouquit, Y.; Craescu, C.T.; Bellissent-Funel, M.-C. Biophysical Study of Thermal Denaturation of Apo-Calmodulin: Dynamics of Native and Unfolded States. Biophys. J. 2008, 95, 5247–5256. [Google Scholar] [CrossRef] [Green Version]
- Henglein, A.J.W.T.; Spinks, R.J. Woods: An Introduction to Radiation Chemistry, Third Edition, John-Wiley and Sons, Inc., New York, Toronto 1990. ISBN 0-471-61403-3. 574 Seiten, Preis: DM 91, 45. Berichte Bunsenges. Phys. Chem. 1991, 95, 451. [Google Scholar] [CrossRef]
- Schägger, H.; von Jagow, G. Tricine-Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis for the Separation of Proteins in the Range from 1 to 100 KDa. Anal. Biochem. 1987, 166, 368–379. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatin, A.; Duchambon, P.; Rest, G.v.d.; Billault, I.; Sicard-Roselli, C. Protein Dimerization via Tyr Residues: Highlight of a Slow Process with Co-Existence of Numerous Intermediates and Final Products. Int. J. Mol. Sci. 2022, 23, 1174. https://doi.org/10.3390/ijms23031174
Gatin A, Duchambon P, Rest Gvd, Billault I, Sicard-Roselli C. Protein Dimerization via Tyr Residues: Highlight of a Slow Process with Co-Existence of Numerous Intermediates and Final Products. International Journal of Molecular Sciences. 2022; 23(3):1174. https://doi.org/10.3390/ijms23031174
Chicago/Turabian StyleGatin, Anouchka, Patricia Duchambon, Guillaume van der Rest, Isabelle Billault, and Cécile Sicard-Roselli. 2022. "Protein Dimerization via Tyr Residues: Highlight of a Slow Process with Co-Existence of Numerous Intermediates and Final Products" International Journal of Molecular Sciences 23, no. 3: 1174. https://doi.org/10.3390/ijms23031174
APA StyleGatin, A., Duchambon, P., Rest, G. v. d., Billault, I., & Sicard-Roselli, C. (2022). Protein Dimerization via Tyr Residues: Highlight of a Slow Process with Co-Existence of Numerous Intermediates and Final Products. International Journal of Molecular Sciences, 23(3), 1174. https://doi.org/10.3390/ijms23031174