
Renal Cell Cancer and Obesity



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Renal Cell Cancer
3. Impact of Obesity on Renal Cancers
4. Mechanisms Linking Obesity with Cancers
4.1. Hypoxia
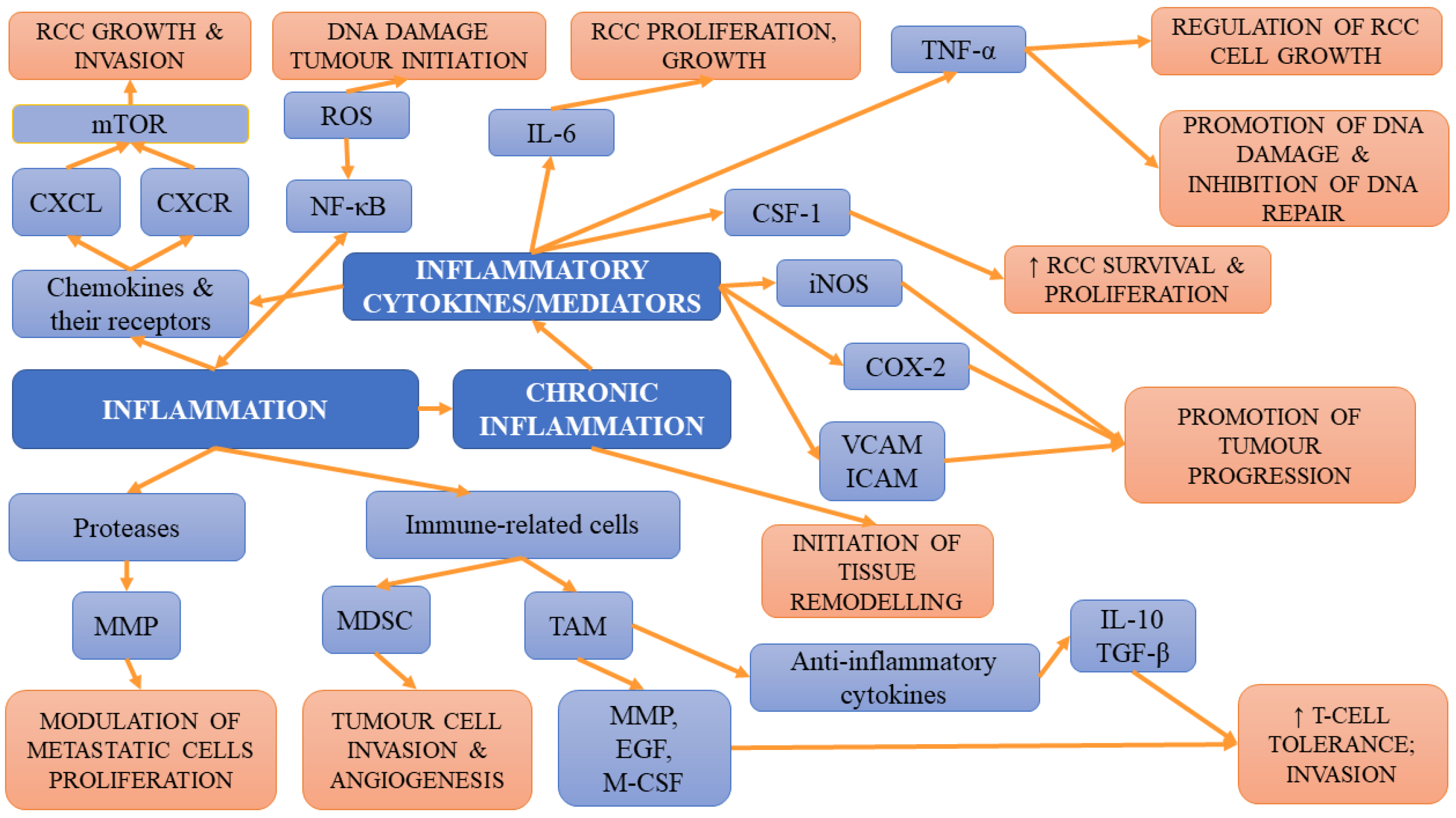
4.2. Inflammation
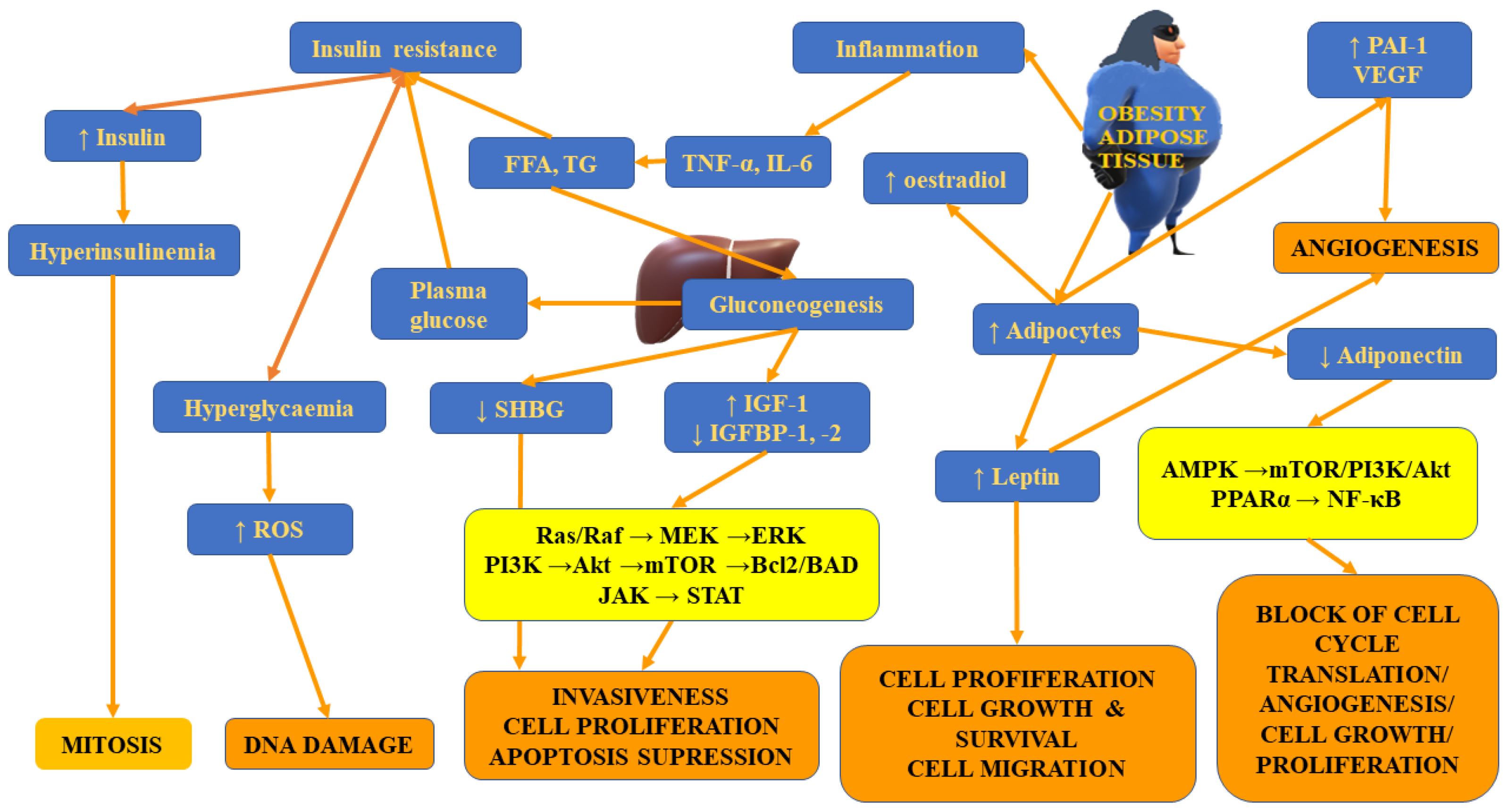
4.3. Insulin Resistance
4.4. Adipokines and Adipose Tissue
4.4.1. Leptin
4.4.2. Adiponectin
4.5. Fatty Acids
4.6. Peroxisome Proliferator-Activated Receptors (PPARs)
4.7. DNA Hypermethylation, miRNAs and Single Nucleotide Polymorphisms (SNPs)
4.8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Huang, X.; Nie, Y.; Shi, X.; Shu, C. A Combined Effect of Expression Levels of Obesity-Related Genes and Clinical Factors on Cancer Survival Rate. BioMed Res. Int. 2020, 2020, 8838676. [Google Scholar] [CrossRef]
- Hursting, S.D.; Nunez, N.P.; Varticovski, L.; Vinson, C. The obesity-cancer link: Lessons learned from a fatless mouse. Cancer Res. 2007, 67, 2391–2393. [Google Scholar] [CrossRef] [Green Version]
- Taubes, G. Unraveling the obesity-cancer connection. Science 2012, 335, 28–32. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, A.; Romano, L.; Di Renzo, L.; Di Lorenzo, N.; Cenname, G.; Gualtieri, P. Obesity: A preventable, treatable, but relapsing disease. Nutrition 2020, 71, 110615. [Google Scholar] [CrossRef]
- Güngör, N.K. Overweight and obesity in children and adolescents. J. Clin. Res. Pediatric. Endocrinol. 2014, 6, 129. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Iizuka, Y.; Kim, H.; Nakasatomi, M.; Izawa, T.; Hirako, S.; Matsumoto, A. Fish oil prevents excessive accumulation of subcutaneous fat caused by an adverse effect of pioglitazone treatment and positively changes adipocytes in KK mice. Toxicol. Rep. 2016, 3, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Unger, R.H. Lipid overload and overflow: Metabolic trauma and the metabolic syndrome. Trends Endocrinol. Metab. 2003, 14, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Park, S.W.; Kim, D.J.; Woo, J. Gender disparity in the secular trends for obesity prevalence in Korea: Analyses based on the KNHANES 1998–2009. Korean J. Intern. Med. 2013, 28, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, P.A.G.; Stevens, G.A.; Ezzati, P.M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Whiteman, D.C.; Wilson, L.F. The fractions of cancer attributable to modifiable factors: A global review. Cancer Epidemiol. 2016, 44, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, L.; Gummesson, A.; Sjöström, C.D.; Narbro, K.; Peltonen, M.; Wedel, H.; Bengtsson, C.; Bouchard, C.; Carlsson, B.; Dahlgren, S.; et al. Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects Study): A prospective, controlled intervention trial. Lancet Oncol. 2009, 10, 653–662. [Google Scholar] [CrossRef]
- Adams, T.D.; Stroup, A.M.; Gress, R.E.; Adams, K.F.; Calle, E.E.; Smith, S.C.; Halverson, R.C.; Simper, S.C.; Hopkins, P.N.; Hunt, S.C. Cancer incidence and mortality after gastric bypass surgery. Obesity 2009, 17, 796–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Li, Q.; Che, X.; Wang, Q.; Wu, G. The Uniqueness of Clear Cell Renal Cell Carcinoma: Summary of the Process and Abnormality of Glucose Metabolism and Lipid Metabolism in ccRCC. Front. Oncol. 2021, 11, 727778. [Google Scholar] [CrossRef]
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef]
- Saito, K.; Arai, E.; Maekawa, K.; Ishikawa, M.; Fujimoto, H.; Taguchi, R.; Matsumoto, K.; Kanai, Y.; Saito, Y. Lipidomic signatures and associated transcriptomic profiles of clear cell renal cell carcinoma. Sci. Rep. 2016, 6, 28932. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.P.; Zou, J.; Xu, Z.Q.; Ruan, J.; Yang, S.D.; Yin, Y.; Mu, H.J. Association of leptin, visfatin, apelin, resistin and adiponectin with clear cell renal cell carcinoma. Oncol. Lett. 2017, 13, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.; Akhtar, M.; Beckwith, B.J.; Bugert, P.; Cooper, C.S.; Delahunt, B.; Eble, J.N.; Fleming, S.; Ljungberg, B.; Medeiros, L.J. The Heidelberg classification of renal cell tumours. J. Pathol. J. Pathol. Soc. Great Br. Irel. 1997, 183, 131–133. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Furberg, H.; Zabor, E.C.; Jacobsen, A.; Schultz, N.; Ciriello, G.; Mikklineni, N.; Fiegoli, B.; Kim, P.H.; Voss, M.H.; et al. An epidemiologic and genomic investigation into the obesity paradox in renal cell carcinoma. J. Natl. Cancer Inst. 2013, 105, 1862–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Cairns, P. Renal cell carcinoma. Cancer Biomark. 2011, 9, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Axwijk, P.H.; Kluijt, I.; De Jong, D.; Gille, H.; Teertstra, J.; Horenblas, S. Hereditary causes of kidney tumours. Eur. J. Clin. Investig. 2010, 40, 433–439. [Google Scholar] [CrossRef]
- Christensen, M.B.; Wadt, K.; Jensen, U.B.; Lautrup, C.K.; Bojesen, A.; Krogh, L.N.; Overeem Hansen, T.V.; Gerdes, A.M. Exploring the hereditary background of renal cancer in Denmark. PLoS ONE 2019, 14, e0215725. [Google Scholar] [CrossRef] [Green Version]
- Razafinjatovo, C.; Bihr, S.; Mischo, A.; Vogl, U.; Schmidinger, M.; Moch, H.; Schraml, P. Characterization of VHL missense mutations in sporadic clear cell renal cell carcinoma: Hotspots, affected binding domains, functional impact on pVHL and therapeutic relevance. BMC Cancer 2016, 16, 638. [Google Scholar] [CrossRef] [Green Version]
- Linehan, W.M.; Ricketts, C.J. The Cancer Genome Atlas of renal cell carcinoma: Findings and clinical implications. Nat. Rev. Urol. 2019, 16, 539–552. [Google Scholar] [CrossRef]
- Bergström, A.; Hsieh, C.C.; Lindblad, P.; Lu, C.M.; Cook, N.R.; Wolk, A. Obesity and renal cell cancer—A quantitative review. Br. J. Cancer 2001, 85, 984–990. [Google Scholar] [CrossRef] [Green Version]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, Z.; Wu, X.; Zhang, J.; Zhai, W.; Chen, Y. Identification of genes that correlate clear cell renal cell carcinoma and obesity and exhibit potential prognostic value. Transl. Androl. Urol. 2021, 10, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Massari, F.; Mollica, V.; Rizzo, A.; Cosmai, L.; Rizzo, M.; Porta, C. Safety evaluation of immune-based combinations in patients with advanced renal cell carcinoma: A systematic review and meta-analysis. Expert Opin. Drug Saf. 2020, 19, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Massari, F.; Rizzo, A.; Mollica, V.; Rosellini, M.; Marchetti, A.; Ardizzoni, A.; Santoni, M. Immune-based combinations for the treatment of metastatic renal cell carcinoma: A meta-analysis of randomised clinical trials. Eur. J. Cancer 2021, 154, 120–127. [Google Scholar] [CrossRef]
- Janzen, N.K.; Kim, H.L.; Figlin, R.A.; Belldegrun, A.S. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol. Clin. N. Am. 2003, 30, 843–852. [Google Scholar] [CrossRef]
- Howlader, N.N.; Noone, A.; Krapcho, M.; Garshell, J.; Neyman, N.; Altekruse, S.F.; Kosary, C.L.; Tatalovich, Z.; Cho, H.; Mariotto, A.; et al. Cronin Ke: SEER Cancer Statistics Review, 1975–2010; National Cancer Institute: Bethesda, MD, USA, 2013. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerschbaum, E.; Nüssler, V. Cancer Prevention with Nutrition and Lifestyle. Visc. Med. 2019, 35, 204–209. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer—Mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.L.; Dive, C.; Renehan, A.G. Biological mechanisms linking obesity and cancer risk: New perspectives. Annu. Rev. Med. 2010, 61, 301–316. [Google Scholar] [CrossRef]
- Klinghoffer, Z.; Yang, B.; Kapoor, A.; Pinthus, J.H. Obesity and renal cell carcinoma: Epidemiology, underlying mechanisms and management considerations. Expert Rev. Anticancer Ther. 2009, 9, 975–987. [Google Scholar] [CrossRef]
- Vucenik, I.; Stains, J.P. Obesity and cancer risk: Evidence, mechanisms, and recommendations. Ann. N. Y. Acad. Sci. 2012, 1271, 37. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, F.; Kaaks, R.; Vainio, H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002, 3, 565–574. [Google Scholar] [CrossRef]
- Zhang, G.-M.; Zhu, Y.; Ye, D.-W. Metabolic syndrome and renal cell carcinoma. World J. Surg. Oncol. 2014, 12, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, K.F.; Leitzmann, M.F.; Albanes, D.; Kipnis, V.; Moore, S.C.; Schatzkin, A.; Chow, W.-H. Body Size and Renal Cell Cancer Incidence in a Large US Cohort Study. Am. J. Epidemiol. 2008, 168, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Häggström, C.; Rapp, K.; Stocks, T.; Manjer, J.; Bjørge, T.; Ulmer, H.; Engeland, A.; Almqvist, M.; Concin, H.; Selmer, R.; et al. Metabolic Factors Associated with Risk of Renal Cell Carcinoma. PLoS ONE 2013, 8, e57475. [Google Scholar] [CrossRef]
- Budny, A.; Grochowski, C.; Kozłowski, P.; Kolak, A.; Kamińska, M.; Budny, B.; Abramiuk, M.; Burdan, F. Obesity as a tumour development triggering factor. Ann. Agric. Environ. Med. 2019, 26, 13–23. [Google Scholar] [CrossRef]
- Li, M.; Bu, R. Biological Support to Obesity Paradox in Renal Cell Carcinoma: A Review. Urol. Int. 2020, 104, 837–848. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Aurilio, G.; Piva, F.; Santoni, M.; Cimadamore, A.; Sorgentoni, G.; Lopez-Beltran, A.; Cheng, L.; Battelli, N.; Nolè, F.; Montironi, R. The Role of Obesity in Renal Cell Carcinoma Patients: Clinical-Pathological Implications. Int. J. Mol. Sci. 2019, 20, 5683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, H.-K.; Zhang, H.-L.; Yao, X.-D.; Zhang, S.-L.; Dai, B.; Shen, Y.-J.; Liu, X.-H.; Zhou, L.-P.; Ye, D.-W. Visceral obesity and risk of high grade disease in clinical t1a renal cell carcinoma. J. Urol. 2013, 189, 447–453. [Google Scholar] [CrossRef]
- Turco, F.; Tucci, M.; Di Stefano, R.F.; Samuelly, A.; Bungaro, M.; Audisio, M.; Pisano, C.; Di Maio, M.; Scagliotti, G.V.; Buttigliero, C. Renal cell carcinoma (RCC): Fatter is better? A review on the role of obesity in RCC. Endocr.-Relat. Cancer 2021, 28, R207–R216. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, A.R.; Bates, T. Obesity and breast cancer: A review of the literature. Breast 2004, 13, 85–92. [Google Scholar] [CrossRef]
- Choi, Y.; Park, B.; Jeong, B.C.; Seo, S.I.; Jeon, S.S.; Choi, H.Y.; Adami, H.O.; Lee, J.E.; Lee, H.M. Body mass index and survival in patients with renal cell carcinoma: A clinical-based cohort and meta-analysis. Int. J. Cancer 2013, 132, 625–634. [Google Scholar] [CrossRef]
- Albiges, L.; Hakimi, A.A.; Xie, W.; McKay, R.R.; Simantov, R.; Lin, X.; Lee, J.-L.; Rini, B.I.; Srinivas, S.; Bjarnason, G.A.; et al. Body Mass Index and Metastatic Renal Cell Carcinoma: Clinical and Biological Correlations. J. Clin. Oncol. 2016, 34, 3655–3663. [Google Scholar] [CrossRef]
- Parker, A.S.; Lohse, C.M.; Cheville, J.C.; Thiel, D.D.; Leibovich, B.C.; Blute, M.L. Greater body mass index is associated with better pathologic features and improved outcome among patients treated surgically for clear cell renal cell carcinoma. Urology 2006, 68, 741–746. [Google Scholar] [CrossRef]
- Steffens, S.; Grünwald, V.; Ringe, K.I.; Seidel, C.; Eggers, H.; Schrader, M.; Wacker, F.; Kuczyk, M.A.; Schrader, A.J. Does obesity influence the prognosis of metastatic renal cell carcinoma in patients treated with vascular endothelial growth factor–targeted therapy? Oncologist 2011, 16, 1565. [Google Scholar] [CrossRef] [Green Version]
- Kamat, A.M.; Shock, R.P.; Naya, Y.; Rosser, C.J.; Slaton, J.W.; Pisters, L.L. Prognostic value of body mass index in patients undergoing nephrectomy for localized renal tumors. Urology 2004, 63, 46–50. [Google Scholar] [CrossRef]
- Oreopoulos, A.; Padwal, R.; Kalantar-Zadeh, K.; Fonarow, G.C.; Norris, C.M.; McAlister, F.A. Body mass index and mortality in heart failure: A meta-analysis. Am. Heart J. 2008, 156, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovesdy, C.P.; Anderson, J.E. Cardiovascular and survival paradoxes in dialysis patients: Reverse epidemiology in patients with chronic kidney disease who are not yet on dialysis. Semin. Dial. 2007, 20, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Waalkes, S.; Merseburger, A.S.; Kramer, M.W.; Herrmann, T.R.; Wegener, G.; Rustemeier, J.; Hofmann, R.; Schrader, M.; Kuczyk, M.A.; Schrader, A.J. Obesity is associated with improved survival in patients with organ-confined clear-cell kidney cancer. Cancer Causes Control 2010, 21, 1905–1910. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Furberg, H.; Kuo, F.; Vuong, L.; Ged, Y.; Patil, S.; Ostrovnaya, I.; Petruzella, S.; Reising, A.; Patel, P.; et al. Transcriptomic signatures related to the obesity paradox in patients with clear cell renal cell carcinoma: A cohort study. Lancet Oncol. 2020, 21, 283–293. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Voss, M.H.; Kuo, F.; Sanchez, A.; Liu, M.; Nixon, B.G.; Vuong, L.; Ostrovnaya, I.; Chen, Y.B.; Reuter, V.; et al. Transcriptomic Profiling of the Tumor Microenvironment Reveals Distinct Subgroups of Clear Cell Renal Cell Cancer: Data from a Randomized Phase III Trial. Cancer Discov. 2019, 9, 510–525. [Google Scholar] [CrossRef] [Green Version]
- Haake, S.M.; Brooks, S.A.; Welsh, E.; Fulp, W.J.; Chen, D.-T.; Dhillon, J.; Haura, E.; Sexton, W.; Spiess, P.E.; Pow-Sang, J.; et al. Patients with ClearCode34-identified molecular subtypes of clear cell renal cell carcinoma represent unique populations with distinct comorbidities. Urol. Oncol. 2016, 34, 122.e1–122.e7. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K.M.; Cho, E. Obesity and Kidney Cancer. In Obesity and Cancer; Springer: Cham, Switzerland, 2016; Volume 208, pp. 81–93. [Google Scholar] [CrossRef]
- Cao, Y. Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat. Rev. Drug Discov. 2010, 9, 107–115. [Google Scholar] [CrossRef]
- Lawler, H.M.; Underkofler, C.M.; Kern, P.A.; Erickson, C.; Bredbeck, B.; Rasouli, N. Adipose tissue hypoxia, inflammation, and fibrosis in obese insulin-sensitive and obese insulin-resistant subjects. J. Clin. Endocrinol. Metab. 2016, 101, 1422–1428. [Google Scholar] [CrossRef] [Green Version]
- Challapalli, A.; Carroll, L.; Aboagye, E.O. Molecular mechanisms of hypoxia in cancer. Clin. Transl. Imaging 2017, 5, 225–253. [Google Scholar] [CrossRef] [Green Version]
- Thews, O.; Gassner, B.; Kelleher, D.K.; Schwerd, G.; Gekle, M. Impact of extracellular acidity on the activity of P-glycoprotein and the cytotoxicity of chemotherapeutic drugs. Neoplasia 2006, 8, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Tordjman, J.; Clément, K.; Scherer, P.E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Vegiopoulos, A.; Rohm, M.; Herzig, S. Adipose tissue: Between the extremes. EMBO J. 2017, 36, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzarà, A.; Pirillo, C.; Giovannini, C.; Federico, G.; Scarpato, R. Different repair kinetic of DSBs induced by mitomycin C in peripheral lymphocytes of obese and normal weight adolescents. Mutat. Res. 2016, 789, 9–14. [Google Scholar] [CrossRef]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Rausch, M.; Weisberg, S.; Vardhana, P.; Tortoriello, D. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int. J. Obes. 2008, 32, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Hoefflin, R.; Harlander, S.; Schäfer, S.; Metzger, P.; Kuo, F.; Schönenberger, D.; Adlesic, M.; Peighambari, A.; Seidel, P.; Chen, C.Y.; et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 2020, 11, 4111. [Google Scholar] [CrossRef]
- Chappell, J.C.; Payne, L.B.; Rathmell, W.K. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J. Clin. Investig. 2019, 129, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Liu, P.; Pan, W.; Singh, S.R.; Wei, Y. Hypoxia and hypoxia inducible factors in tumor metabolism. Cancer Lett. 2015, 356, 263–267. [Google Scholar] [CrossRef]
- Tafani, M.; Pucci, B.; Russo, A.; Schito, L.; Pellegrini, L.; Perrone, G.A.; Villanova, L.; Salvatori, L.; Ravenna, L.; Petrangeli, E.; et al. Modulators of HIF1α and NFkB in Cancer Treatment: Is it a Rational Approach for Controlling Malignant Progression? Front. Pharmacol. 2013, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y.; Hamada, J.; Kobayashi, C.; Nakamura, R.; Suzuki, Y.; Kimata, R.; Nishimura, T.; Kitagawa, T.; Kunimoto, M.; Imura, N.; et al. Over expression of hypoxia-inducible factor-1alpha in renal and bladder cancer cells increases tumorigenic potency. J. Urol. 2005, 173, 1762–1766. [Google Scholar] [CrossRef]
- Lidgren, A.; Hedberg, Y.; Grankvist, K.; Rasmuson, T.; Vasko, J.; Ljungberg, B. The expression of hypoxia-inducible factor 1α is a favorable independent prognostic factor in renal cell carcinoma. Clin. Cancer Res. 2005, 11, 1129–1135. [Google Scholar] [PubMed]
- Klatte, T.; Seligson, D.B.; Riggs, S.B.; Leppert, J.T.; Berkman, M.K.; Kleid, M.D.; Yu, H.; Kabbinavar, F.F.; Pantuck, A.J.; Belldegrun, A.S. Hypoxia-inducible factor 1 alpha in clear cell renal cell carcinoma. Clin. Cancer Res. 2007, 13, 7388–7393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Rettig, M.B. Mechanism of von Hippel-Lindau protein-mediated suppression of nuclear factor kappa B activity. Mol. Cell. Biol. 2005, 25, 7546–7556. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.-K.; Heo, Y.; Lee, H.S.; Lee, M.Y. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Gordan, J.D.; Lal, P.; Dondeti, V.R.; Letrero, R.; Parekh, K.N.; Oquendo, C.E.; Greenberg, R.A.; Flaherty, K.T.; Rathmell, W.K.; Keith, B.; et al. HIF-α effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 2008, 14, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Beroukhim, R.; Schumacher, S.E.; Zhou, J.; Chang, M.; Signoretti, S.; Kaelin, W.G. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov. 2011, 1, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.-L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef] [Green Version]
- Mandriota, S.J.; Turner, K.J.; Davies, D.R.; Murray, P.G.; Morgan, N.V.; Sowter, H.M.; Wykoff, C.C.; Maher, E.R.; Harris, A.L.; Ratcliffe, P.J.; et al. HIF activation identifies early lesions in VHL kidneys: Evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 2002, 1, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Gudas, L.J.; Fu, L.; Minton, D.R.; Mongan, N.P.; Nanus, D.M. The role of HIF1α in renal cell carcinoma tumorigenesis. J. Mol. Med. 2014, 92, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Wang, G.; Shevchuk, M.M.; Nanus, D.M.; Gudas, L.J. Activation of HIF2α in kidney proximal tubule cells causes abnormal glycogen deposition but not tumorigenesis. Cancer Res. 2013, 73, 2916–2925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönenberger, D.; Harlander, S.; Rajski, M.; Jacobs, R.A.; Lundby, A.-K.; Adlesic, M.; Hejhal, T.; Wild, P.J.; Lundby, C.; Frew, I.J. Formation of renal cysts and tumors in Vhl/Trp53-deficient mice requires HIF1α and HIF2α. Cancer Res. 2016, 76, 2025–2036. [Google Scholar] [CrossRef] [Green Version]
- Brestoff, J.R.; Artis, D. Immune Regulation of Metabolic Homeostasis in Health and Disease. Cell 2015, 161, 146–160. [Google Scholar] [CrossRef] [Green Version]
- Maihöfner, C.; Charalambous, M.P.; Bhambra, U.; Lightfoot, T.; Geisslinger, G.; Gooderham, N.J.; Colorectal Cancer Group. Expression of cyclooxygenase-2 parallels expression of interleukin-1beta, interleukin-6 and NF-kappaB in human colorectal cancer. Carcinogenesis 2003, 24, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Osório-Costa, F.; Carvalheira, J.B.C. TNF-α in obesity-associated colon cancer. Transl. Gastrointest. Cancer 2013, 2, 179–193. [Google Scholar]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Dudzinski, S.O.; Bader, J.E.; Beckermann, K.E.; Young, K.L.; Hongo, R.; Madden, M.Z.; Abraham, A.; Reinfeld, B.E.; Ye, X.; MacIver, N.J.; et al. Leptin Augments Antitumor Immunity in Obesity by Repolarizing Tumor-Associated Macrophages. J. Immunol. 2021, 207, 3122–3130. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 2017, 47, 406–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. 2016, 11, 421–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, C.L.; Lysaght, J.; O’Sullivan, J.; Reynolds, J.V. Emerging Concepts Linking Obesity with the Hallmarks of Cancer. Trends Endocrinol. Metab. 2017, 28, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Solá, E.; Jover, A.; López-Ruiz, A.; Jarabo, M.; Vayá, A.; Morillas, C.; Gómez-Balaguer, M.; Hernández-Mijares, A. Parameters of inflammation in morbid obesity: Lack of effect of moderate weight loss. Obes. Surg. 2009, 19, 571–576. [Google Scholar] [CrossRef]
- Kuo, S.-H.; Yang, S.-Y.; You, S.-L.; Lien, H.-C.; Lin, C.-H.; Lin, P.-H.; Huang, C.-S. Polymorphisms of ESR1, UGT1A1, HCN1, MAP3K1 and CYP2B6 are associated with the prognosis of hormone receptor-positive early breast cancer. Oncotarget 2017, 8, 20925–20938. [Google Scholar] [CrossRef]
- Rethorst, C.D.; Bernstein, I.; Trivedi, M.H. Inflammation, obesity, and metabolic syndrome in depression: Analysis of the 2009-2010 National Health and Nutrition Examination Survey (NHANES). J. Clin. Psychiatry 2014, 75, e1428–e1432. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Yang, Q.; Cao, J.; Xie, N.; Liu, K.; Shou, P.; Qian, F.; Wang, Y.; Shi, Y. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis. 2016, 7, e2167. [Google Scholar] [CrossRef] [Green Version]
- Incio, J.; Tam, J.; Rahbari, N.N.; Suboj, P.; McManus, D.T.; Chin, S.M.; Vardam, T.D.; Batista, A.; Babykutty, S.; Jung, K.; et al. PlGF/VEGFR-1 signaling promotes macrophage polarization and accelerated tumor progression in obesity. Clin. Cancer Res. 2016, 22, 2993–3004. [Google Scholar] [CrossRef] [Green Version]
- Ramkhelawon, B.; Hennessy, E.J.; Ménager, M.; Ray, T.D.; Sheedy, F.J.; Hutchison, S.; Wanschel, A.; Oldebeken, S.; Geoffrion, M.; Spiro, W.; et al. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat. Med. 2014, 20, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Morinaga, H.; Talukdar, S.; Bae, E.J.; Olefsky, J.M. Increased macrophage migration into adipose tissue in obese mice. Diabetes 2012, 61, 346–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springer, N.L.; Iyengar, N.M.; Bareja, R.; Verma, A.; Jochelson, M.S.; Giri, D.D.; Zhou, X.K.; Elemento, O.; Dannenberg, A.J.; Fischbach, C. Obesity-Associated Extracellular Matrix Remodeling Promotes a Macrophage Phenotype Similar to Tumor-Associated Macrophages. Am. J. Pathol. 2019, 189, 2019–2035. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R.; Subbaramaiah, K.; Hudis, C.A.; Dannenberg, A.J. Molecular pathways: Adipose inflammation as a mediator of obesity-associated cancer. Clin. Cancer Res. 2013, 19, 6074–6083. [Google Scholar] [CrossRef] [Green Version]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [Green Version]
- Malekghasemi, S.; Majidi, J.; Baghbanzadeh, A.; Abdolalizadeh, J.; Baradaran, B.; Aghebati-Maleki, L. Tumor-Associated Macrophages: Protumoral Macrophages in Inflammatory Tumor Microenvironment. Adv. Pharm. Bull. 2020, 10, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.; Warren, G.; Wei, X. Macrophages associated with tumors as potential targets and therapeutic intermediates. Nanomedicine 2014, 9, 695–707. [Google Scholar] [CrossRef] [Green Version]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- Cardillo, M.R.; Ippoliti, F. Interleukin-6, interleukin-10 and heat shock protein-90 expression in renal epithelial neoplasias and surrounding normal-appearing renal parenchyma. Int. J. Immunopathol. Pharmacol. 2007, 20, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Ho, M.-Y.; Tang, S.-J.; Chuang, M.-J.; Cha, T.-L.; Li, J.-Y.; Sun, G.-H.; Sun, K.-H. TNF-α induces epithelial–mesenchymal transition of renal cell carcinoma cells via a GSK3β-dependent mechanism. Mol. Cancer Res. 2012, 10, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.; Cao, L.; Zhou, H. Expression of COX-2 and VEGF and their correlation with angiogenesis in human clear cell renal cell carcinoma. Zhonghua Zhong Liu Za Zhi [Chin. J. Oncol.] 2009, 31, 687–690. [Google Scholar] [PubMed]
- Sun, H.; Wang, H.; Qin, W.; Yang, B.; Wang, S.; Jian, B. Expression of IGF-IR and COX-2 in renal cell carcinoma and their relationship with cell proliferation. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi [Chin. J. Cell. Mol. Immunol.] 2009, 25, 348–350. [Google Scholar]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Gentile, P.; Kim, B.-S.; Cervelli, V.; Orlandi, A. Adipose-Derived Stem Cells in Cancer Progression: New Perspectives and Opportunities. Int. J. Mol. Sci. 2019, 20, 3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westley, R.L.; May, F.E.B. A Twenty-First Century Cancer Epidemic Caused by Obesity: The Involvement of Insulin, Diabetes, and Insulin-Like Growth Factors. Int. J. Endocrinol. 2013, 2013, 632461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, P.A.; Ranganathan, S.; Li, C.; Wood, L.; Ranganathan, G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E745–E751. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.A.; Saghizadeh, M.; Ong, J.M.; Bosch, R.J.; Deem, R.; Simsolo, R.B. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J. Clin. Investig. 1995, 95, 2111–2119. [Google Scholar] [CrossRef] [Green Version]
- Pasquali, R.; Casimirri, F.; De Iasio, R.; Mesini, P.; Boschi, S.; Chierici, R.; Flamia, R.; Biscotti, M.; Vicennati, V. Insulin regulates testosterone and sex hormone-binding globulin concentrations in adult normal weight and obese men. J. Clin. Endocrinol. Metab. 1995, 80, 654–658. [Google Scholar] [CrossRef]
- Simó, R.; Saez-Lopez, C.; Lecube, A.; Hernandez, C.; Fort, J.M.; Selva, D.M. Adiponectin upregulates SHBG production: Molecular mechanisms and potential implications. Endocrinology 2014, 155, 2820–2830. [Google Scholar] [CrossRef] [Green Version]
- Weaver, J.U.; Holly, J.M.P.; Kopelman, P.G.; Noonan, K.; Giadom, C.G.; White, N.; Virdee, S.; Wass, J.A.H. Decreased sex hormone binding globulin (shbg) and insulin-like growth factor binding protein (igfbp-1) in extreme obesity. Clin. Endocrinol. 1990, 33, 415–422. [Google Scholar] [CrossRef]
- Sciacca, L.; Cassarino, M.F.; Genua, M.; Vigneri, P.; Giovanna Pennisi, M.; Malandrino, P.; Squatrito, S.; Pezzino, V.; Vigneri, R. Biological effects of insulin and its analogs on cancer cells with different insulin family receptor expression. J. Cell. Physiol. 2014, 229, 1817–1821. [Google Scholar] [CrossRef]
- Matyszewski, A.; Czarnecka, A.; Kawecki, M.; Korzeń, P.; Safir, I.J.; Kukwa, W.; Szczylik, C. Impaired glucose metabolism treatment and carcinogenesis. Oncol. Lett. 2015, 10, 589–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuveni, H.; Flashner-Abramson, E.; Steiner, L.; Makedonski, K.; Song, R.; Shir, A.; Herlyn, M.; Bar-Eli, M.; Levitzki, A. Therapeutic destruction of insulin receptor substrates for cancer treatment. Cancer Res. 2013, 73, 4383–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solarek, W.; Koper, M.; Lewicki, S.; Szczylik, C.; Czarnecka, A.M. Insulin and insulin-like growth factors act as renal cell cancer intratumoral regulators. J. Cell Commun. Signal. 2019, 13, 381–394. [Google Scholar] [CrossRef] [Green Version]
- Gatica, R.; Bertinat, R.; Silva, P.; Carpio, D.; Ramírez, M.J.; Slebe, J.C.; Martín, R.S.; Nualart, F.; Campistol, J.M.; Caelles, C.; et al. Altered expression and localization of insulin receptor in proximal tubule cells from human and rat diabetic kidney. J. Cell. Biochem. 2013, 114, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Inoue, T.; Huang, M.; Numakura, K.; Tsuruta, H.; Saito, M.; Maeno, A.; Nakamura, E.; Narita, S.; Tsuchiya, N.; et al. Inverse relationship between insulin receptor expression and progression in renal cell carcinoma. Oncol. Rep. 2017, 37, 2929–2941. [Google Scholar] [CrossRef] [Green Version]
- Ougolkov, A.V.; Bone, N.D.; Fernandez-Zapico, M.E.; Kay, N.E.; Billadeau, D.D. Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor κB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood 2007, 110, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Schips, L.; Zigeuner, R.; Ratschek, M.; Rehak, P.; Rüschoff, J.; Langner, C. Analysis of insulin-like growth factors and insulin-like growth factor I receptor expression in renal cell carcinoma. Am. J. Clin. Pathol. 2004, 122, 931–937. [Google Scholar] [CrossRef]
- Cai, W.; Sakaguchi, M.; Kleinridders, A.; Gonzalez-Del Pino, G.; Dreyfuss, J.M.; O’Neill, B.T.; Ramirez, A.K.; Pan, H.; Winnay, J.N.; Boucher, J.; et al. Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat. Commun. 2017, 8, 14892. [Google Scholar] [CrossRef]
- Cardillo, T.M.; Trisal, P.; Arrojo, R.; Goldenberg, D.M.; Chang, C.-H. Targeting both IGF-1R and mTOR synergistically inhibits growth of renal cell carcinoma in vitro. BMC Cancer 2013, 13, 170. [Google Scholar] [CrossRef] [Green Version]
- Tracz, A.F.; Szczylik, C.; Porta, C.; Czarnecka, A.M. Insulin-like growth factor-1 signaling in renal cell carcinoma. BMC Cancer 2016, 16, 453. [Google Scholar] [CrossRef] [Green Version]
- Rasmuson, T.; Grankvist, K.; Jacobsen, J.; Olsson, T.; Ljungberg, B. Serum insulin-like growth factor-1 is an independent predictor of prognosis in patients with renal cell carcinoma. Acta Oncol. 2004, 43, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; Yee, D. Insulin-like growth factor-I and cancer risk. Growth Horm. IGF Res. 2004, 14, 261–269. [Google Scholar] [CrossRef]
- Chuang, S.-T.; Patton, K.T.; Schafernak, K.T.; Papavero, V.; Lin, F.; Baxter, R.C.; Teh, B.T.; Yang, X.J. Over expression of insulin-like growth factor binding protein 3 in clear cell renal cell carcinoma. J. Urol. 2008, 179, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Perseghin, G.; Calori, G.; Lattuada, G.; Ragogna, F.; Dugnani, E.; Garancini, M.P.; Crosignani, P.; Villa, M.; Bosi, E.; Ruotolo, G.; et al. Insulin resistance/hyperinsulinemia and cancer mortality: The Cremona study at the 15th year of follow-up. Acta Diabetol. 2012, 49, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Van Kruijsdijk, R.C.; Van Der Wall, E.; Visseren, F.L. Obesity and cancer: The role of dysfunctional adipose tissue. Cancer Epidemiol. Prev. Biomark. 2009, 18, 2569–2578. [Google Scholar] [CrossRef] [Green Version]
- Blee, A.M.; Huang, H. Fat lure: Adipocytes attract cancer cells out of the prostate. Transl. Cancer Res. 2016, 5, S123–S125. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, R.J.; Monteiro, C.P.; Cunha, V.F.; Azevedo, A.S.; Oliveira, M.J.; Monteiro, R.; Fraga, A.M.; Príncipe, P.; Lobato, C.; Lobo, F.; et al. Tumor cell-educated periprostatic adipose tissue acquires an aggressive cancer-promoting secretory profile. Cell. Physiol. Biochem. 2012, 29, 233–240. [Google Scholar] [CrossRef]
- Zhang, Y.; Daquinag, A.C.; Amaya-Manzanares, F.; Sirin, O.; Tseng, C.; Kolonin, M.G. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res. 2012, 72, 5198–5208. [Google Scholar] [CrossRef] [Green Version]
- Margetic, S.; Gazzola, C.; Pegg, G.; Hill, R. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum Immunoreactive-Leptin Concentrations in Normal-Weight and Obese Humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, A.; Sumitomo, M.; Asakuma, J.; Asano, T.; Zheng, R.; Asano, T.; Nanus, D.M.; Hayakawa, M. Increased serum leptin levels and over expression of leptin receptors are associated with the invasion and progression of renal cell carcinoma. J. Urol. 2006, 176, 1631–1635. [Google Scholar] [CrossRef] [PubMed]
- Spyridopoulos, T.N.; Petridou, E.T.; Dessypris, N.; Terzidis, A.; Skalkidou, A.; Deliveliotis, C.; Chrousos, G.P. Inverse association of leptin levels with renal cell carcinoma: Results from a case-control study. Hormones 2009, 8, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiguchi, A.; Sumitomo, M.; Asakuma, J.; Asano, T.; Zheng, R.; Asano, T.; Nanus, D.M.; Hayakawa, M. Leptin promotes invasiveness of murine renal cancer cells via extracellular signal-regulated kinases and rho dependent pathway. J. Urol. 2006, 176, 1636–1641. [Google Scholar] [CrossRef]
- Shimizu, H.; Mori, M. Role of leptin and its receptor in the regulation of appetite and body fat. Nihon Rinsho. Jpn. J. Clin. Med. 2001, 59, 421–426. [Google Scholar]
- Gonzalez-Perez, R.R.; Xu, Y.; Guo, S.; Watters, A.; Zhou, W.; Leibovich, S.J. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFκB/HIF-1α activation. Cell. Signal. 2010, 22, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chang, Y.C.; Lan, M.S.; Breslin, M. Leptin stimulates ovarian cancer cell growth and inhibits apoptosis by increasing cyclin D1 and Mcl-1 expression via the activation of the MEK/ERK1/2 and PI3K/Akt signaling pathways. Int. J. Oncol. 2013, 42, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef]
- Rose, D.P.; Komninou, D.; Stephenson, G.D. Obesity, adipocytokines, and insulin resistance in breast cancer. Obes. Rev. 2004, 5, 153–165. [Google Scholar] [CrossRef]
- Li, L.; Gao, Y.; Zhang, L.-L.; He, D.-l. Concomitant activation of the JAK/STAT3 and ERK1/2 signaling is involved in leptin-mediated proliferation of renal cell carcinoma Caki-2 cells. Cancer Biol. Ther. 2008, 7, 1787–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teoh, S.L.; Das, S. Tumour biology of obesity-related cancers: Understanding the molecular concept for better diagnosis and treatment. Tumor Biol. 2016, 37, 14363–14380. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, A.; Marfella, R.; Coppola, L.; Tagliamonte, E.; Fontana, D.; Liguori, E.; Cirillo, T.; Cafiero, M.; Natale, S.; Astarita, C. Effect of weight loss on coronary circulation and adiponectin levels in obese women. Int. J. Cardiol. 2009, 134, 414–416. [Google Scholar] [CrossRef]
- Spyridopoulos, T.N.; Petridou, E.T.; Skalkidou, A.; Dessypris, N.; Chrousos, G.P.; Mantzoros, C.S.; Obesity and Cancer Oncology Group. Low adiponectin levels are associated with renal cell carcinoma: A case-control study. Int. J. Cancer 2007, 120, 1573–1578. [Google Scholar] [CrossRef]
- Sugiyama, M.; Takahashi, H.; Hosono, K.; Endo, H.; Kato, S.; Yoneda, K.; Nozaki, Y.; Fujita, K.; Yoneda, M.; Wada, K.; et al. Adiponectin inhibits colorectal cancer cell growth through the AMPK/mTOR pathway. Int. J. Oncol. 2009, 34, 339–344. [Google Scholar]
- Renehan, A.G.; Zwahlen, M.; Egger, M. Adiposity and cancer risk: New mechanistic insights from epidemiology. Nat. Rev. Cancer 2015, 15, 484–498. [Google Scholar] [CrossRef]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef] [Green Version]
- Kamada, Y.; Matsumoto, H.; Tamura, S.; Fukushima, J.; Kiso, S.; Fukui, K.; Igura, T.; Maeda, N.; Kihara, S.; Funahashi, T.; et al. Hypoadiponectinemia accelerates hepatic tumor formation in a nonalcoholic steatohepatitis mouse model. J. Hepatol. 2007, 47, 556–564. [Google Scholar] [CrossRef]
- Pinthus, J.H.; Kleinmann, N.; Tisdale, B.; Chatterjee, S.; Lu, J.-P.; Gillis, A.; Hamlet, T.; Singh, G.; Farrokhyar, F.; Kapoor, A. Lower plasma adiponectin levels are associated with larger tumor size and metastasis in clear-cell carcinoma of the kidney. Eur. Urol. 2008, 54, 866–874. [Google Scholar] [CrossRef]
- Horiguchi, A.; Ito, K.; Sumitomo, M.; Kimura, F.; Asano, T.; Hayakawa, M. Decreased serum adiponectin levels in patients with metastatic renal cell carcinoma. Jpn. J. Clin. Oncol. 2008, 38, 106–111. [Google Scholar] [CrossRef] [PubMed]
- de Martino, M.; Leitner, C.V.; Hofbauer, S.L.; Lucca, I.; Haitel, A.; Shariat, S.F.; Klatte, T. Serum Adiponectin Predicts Cancer-specific Survival of Patients with Renal Cell Carcinoma. Eur. Urol. Focus 2016, 2, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, I.; Kelesidis, T.; Mantzoros, C.S. Adiponectin and cancer: A systematic review. Br. J. Cancer 2006, 94, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Philippova, M.; Joshi, M.B.; Kyriakakis, E.; Pfaff, D.; Erne, P.; Resink, T.J. A guide and guard: The many faces of T-cadherin. Cell. Signal. 2009, 21, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Narita, S.; Huang, M.; Nara, T.; Numakura, K.; Takayama, K.; Tsuruta, H.; Maeno, A.; Saito, M.; Inoue, T.; et al. The impact of obesity and adiponectin signaling in patients with renal cell carcinoma: A potential mechanism for the “obesity paradox”. PLoS ONE 2017, 12, e0171615. [Google Scholar] [CrossRef]
- Gavrila, A.; Peng, C.K.; Chan, J.L.; Mietus, J.E.; Goldberger, A.L.; Mantzoros, C.S. Diurnal and ultradian dynamics of serum adiponectin in healthy men: Comparison with leptin, circulating soluble leptin receptor, and cortisol patterns. J. Clin. Endocrinol. Metab. 2003, 88, 2838–2843. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, R.; Sharma, A.; Kapoor, M.; Sundararajan, K.; Perruccio, A.V. Racial Differences in Serum Adipokine and Insulin Levels in a Matched Osteoarthritis Sample: A Pilot Study. J. Obes. 2016, 2016, 8746268. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, M.E.; Cleary, M.P. The balance between leptin and adiponectin in the control of carcinogenesis—Focus on mammary tumorigenesis. Biochimie 2012, 94, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, K.; Kenny, H.; Penicka, C.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.; Romero, I.; Carey, M.; Mills, G.; Hotamisligil, G.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharpure, K.M.; Pradeep, S.; Sans, M.; Rupaimoole, R.; Ivan, C.; Wu, S.Y.; Bayraktar, E.; Nagaraja, A.S.; Mangala, L.S.; Zhang, X.; et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018, 9, 2923. [Google Scholar] [CrossRef] [Green Version]
- Horiguchi, A.; Asano, T.; Asano, T.; Ito, K.; Sumitomo, M.; Hayakawa, M. Fatty Acid Synthase Over Expression is an Indicator of Tumor Aggressiveness and Poor Prognosis in Renal Cell Carcinoma. J. Urol. 2008, 180, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nosho, K.; Meyerhardt, J.A.; Kirkner, G.J.; Chan, A.T.; Kawasaki, T.; Giovannucci, E.L.; Loda, M.; Fuchs, C.S. Cohort study of fatty acid synthase expression and patient survival in colon cancer. J. Clin. Oncol. 2008, 26, 5713–5720. [Google Scholar] [CrossRef]
- Nguyen, P.L.; Ma, J.; Chavarro, J.E.; Freedman, M.L.; Lis, R.; Fedele, G.; Fiore, C.; Qiu, W.; Fiorentino, M.; Finn, S.; et al. Fatty acid synthase polymorphisms, tumor expression, body mass index, prostate cancer risk, and survival. J. Clin. Oncol. 2010, 28, 3958–3964. [Google Scholar] [CrossRef] [Green Version]
- Horiguchi, A.; Asano, T.; Asano, T.; Ito, K.; Sumitomo, M.; Hayakawa, M. Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells. J. Urol. 2008, 180, 729–736. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Yaghoubizadeh, M.; Pishkar, L.; Basati, G. Aberrant expression of Peroxisome Proliferator-Activated Receptors in colorectal cancer and their association with cancer progression and prognosis. Gastrointest. Tumors 2020, 7, 11–20. [Google Scholar] [CrossRef]
- Inoue, K.; Kawahito, Y.; Tsubouchi, Y.; Kohno, M.; Yoshimura, R.; Yoshikawa, T.; Sano, H. Expression of peroxisome proliferator-activated receptor gamma in renal cell carcinoma and growth inhibition by its agonists. Biochem. Biophys. Res. Commun. 2001, 287, 727–732. [Google Scholar] [CrossRef]
- Yang, F.G.; Zhang, Z.W.; Xin, D.Q.; Shi, C.J.; Wu, J.P.; Guo, Y.L.; Guan, Y.F. Peroxisome proliferator-activated receptor gamma ligands induce cell cycle arrest and apoptosis in human renal carcinoma cell lines. Acta Pharmacol. Sin. 2005, 26, 753–761. [Google Scholar] [CrossRef]
- Fujita, M.; Tohji, C.; Honda, Y.; Yamamoto, Y.; Nakamura, T.; Yagami, T.; Yamamori, M.; Okamura, N. Cytotoxicity of 15-Deoxy-Δ12, 14-prostaglandin J2 through PPARγ-independent Pathway and the Involvement of the JNK and Akt Pathway in Renal Cell Carcinoma. Int. J. Med. Sci. 2012, 9, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Deguchi, Y.; Tian, R.; Wei, D.; Wu, L.; Chen, W.; Xu, W.; Xu, M.; Liu, F.; Gao, S.; et al. Pleiotropic effects of PPARD accelerate colorectal tumorigenesis, progression, and invasion. Cancer Res. 2019, 79, 954–969. [Google Scholar] [CrossRef] [Green Version]
- Zuo, X.; Xu, W.; Xu, M.; Tian, R.; Moussalli, M.J.; Mao, F.; Zheng, X.; Wang, J.; Morris, J.S.; Gagea, M.; et al. Metastasis regulation by PPARD expression in cancer cells. JCI Insight 2017, 2, e91419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, F.; Xu, M.; Zuo, X.; Yu, J.; Xu, W.; Moussalli, M.J.; Elias, E.; Li, H.S.; Watowich, S.S.; Shureiqi, I. 15-Lipoxygenase-1 suppression of colitis-associated colon cancer through inhibition of the IL-6/STAT3 signaling pathway. FASEB J. 2015, 29, 2359–2370. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.S.; Yip, Y.S.; Lim, E.K.Y.; Wahli, W.; Tan, N.S. PPARs and Tumor Microenvironment: The Emerging Roles of the Metabolic Master Regulators in Tumor Stromal-Epithelial Crosstalk and Carcinogenesis. Cancers 2021, 13, 2153. [Google Scholar] [CrossRef]
- Liu, L.; Yang, Z.; Xu, Y.; Li, J.; Xu, D.; Zhang, L.; Sun, J.; Xia, S.; Zou, F.; Liu, Y. Inhibition of oxidative stress-elicited AKT activation facilitates PPARγ agonist-mediated inhibition of stem cell character and tumor growth of liver cancer cells. PLoS ONE 2013, 8, e73038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef]
- Reka, A.K.; Kurapati, H.; Narala, V.R.; Bommer, G.; Chen, J.; Standiford, T.J.; Keshamouni, V.G. Peroxisome proliferator-activated receptor-γ activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol. Cancer Ther. 2010, 9, 3221–3232. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Chu, E.S.; Zhao, G.; Man, K.; Wu, C.W.; Cheng, J.; Li, G.; Nie, Y.; Lo, C.; Teoh, N.; et al. PPARgamma inhibits hepatocellular carcinoma metastases in vitro and in mice. Br. J. Cancer 2012, 106, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Pérez, J.; Gu, J.; Herrera, L.A.; Tannir, N.M.; Zhang, S.; Matin, S.; Karam, J.A.; Wood, C.G.; Wu, X. Prognostic significance of promoter CpG island methylation of obesity-related genes in patients with nonmetastatic renal cell carcinoma. Cancer 2017, 123, 3617–3627. [Google Scholar] [CrossRef] [Green Version]
- Dulaimi, E.; Ibanez de Caceres, I.; Uzzo, R.G.; Al-Saleem, T.; Greenberg, R.E.; Polascik, T.J.; Babb, J.S.; Grizzle, W.E.; Cairns, P. Promoter hypermethylation profile of kidney cancer. Clin. Cancer Res. 2004, 10, 3972–3979. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.Y.; Mohtat, D.; Yu, Y.; Ko, Y.A.; Shenoy, N.; Bhattacharya, S.; Izquierdo, M.C.; Park, A.S.; Giricz, O.; Vallumsetla, N.; et al. Kidney cancer is characterized by aberrant methylation of tissue-specific enhancers that are prognostic for overall survival. Clin. Cancer Res. 2014, 20, 4349–4360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdu Allah, A.M.; El-Hefnway, S.M.; Alhanafy, A.M.; Zahran, A.M.; Kasem, H.E. Leptin receptor gene (A/G) polymorphism rs1137101 and renal cell carcinoma. Mol. Cell. Biochem. 2018, 448, 137–144. [Google Scholar] [CrossRef]
- Shu, X.; Lin, J.; Wood, C.G.; Tannir, N.M.; Wu, X. Energy balance, polymorphisms in the mTOR pathway, and renal cell carcinoma risk. J. Natl. Cancer Inst. 2013, 105, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Gu, C.; Zhu, Y.; Luo, L.; Dong, D.; Wan, F.; Zhang, H.; Shi, G.; Sun, L.; Ye, D. ADIPOQ polymorphism rs182052 is associated with clear cell renal cell carcinoma. Cancer Sci. 2015, 106, 687–691. [Google Scholar] [CrossRef] [Green Version]
- Brennan, P.; McKay, J.; Moore, L.; Zaridze, D.; Mukeria, A.; Szeszenia-Dabrowska, N.; Lissowska, J.; Rudnai, P.; Fabianova, E.; Mates, D.; et al. Obesity and cancer: Mendelian randomization approach utilizing the FTO genotype. Int. J. Epidemiol. 2009, 38, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Shu, X.; Purdue, M.P.; Ye, Y.; Tu, H.; Wood, C.G.; Tannir, N.M.; Wang, Z.; Albanes, D.; Gapstur, S.M.; Stevens, V.L.; et al. Potential Susceptibility Loci Identified for Renal Cell Carcinoma by Targeting Obesity-Related Genes. Cancer Epidemiol. Prev. Biomark. 2017, 26, 1436–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrissey, J.J.; Mobley, J.; Figenshau, R.S.; Vetter, J.; Bhayani, S.; Kharasch, E.D. Urine aquaporin 1 and perilipin 2 differentiate renal carcinomas from other imaged renal masses and bladder and prostate cancer. Mayo Clin. Proc. 2015, 90, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, J.J.; Kharasch, E.D. The specificity of urinary aquaporin 1 and perilipin 2 to screen for renal cell carcinoma. J. Urol. 2013, 189, 1913–1920. [Google Scholar] [CrossRef] [Green Version]
- Butz, H.; Szabó, P.M.; Nofech-Mozes, R.; Rotondo, F.; Kovacs, K.; Mirham, L.; Girgis, H.; Boles, D.; Patocs, A.; Yousef, G.M. Integrative bioinformatics analysis reveals new prognostic biomarkers of clear cell renal cell carcinoma. Clin. Chem. 2014, 60, 1314–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milosevic, V.S.; Vukmirovic, F.C.; Krstic, M.S.; Zindovic, M.M.; Lj Stojanovic, D.; Jancic, S.A. Involvement of leptin receptors expression in proliferation and neoangiogenesis in colorectal carcinoma. J. BUON 2015, 20, 100–108. [Google Scholar] [PubMed]
- Zhang, Y.; Liu, L.; Li, C.; Ai, H. Correlation analysis between the expressions of leptin and its receptor (ObR) and clinicopathology in endometrial cancer. Cancer Biomark. 2014, 14, 353–359. [Google Scholar] [CrossRef]
- Rodrigues, P.R.; Maia, L.L.; Santos, M.; Peterle, G.T.; Alves, L.U.; Takamori, J.T.; Souza, R.P.; Barbosa, W.M.; Mercante, A.M.; Nunes, F.D.; et al. Leptin receptor expression and Gln223Arg polymorphism as prognostic markers in oral and oropharyngeal cancer. Genet. Mol. Res. 2015, 14, 14979–14988. [Google Scholar] [CrossRef]
- Menghi, F.; Orzan, F.N.; Eoli, M.; Farinotti, M.; Maderna, E.; Pisati, F.; Bianchessi, D.; Valletta, L.; Lodrini, S.; Galli, G.; et al. DNA microarray analysis identifies CKS2 and LEPR as potential markers of meningioma recurrence. Oncologist 2011, 16, 1440–1450. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.P.; Liu, C.L.; Hsu, Y.C.; Chang, Y.C.; Huang, S.Y.; Lee, J.J. Regulation of leptin receptor expression in human papillary thyroid cancer cells. BioMed. Pharmacother. 2012, 66, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ali, S.; Ahmad, A.; Bao, B.; Philip, P.; Sarkar, F. Expression of microRNAs: Potential molecular link between obesity, diabetes and cancer. Obes. Rev. 2011, 12, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, X.; Zhang, H.; Xiang, Y.; Chen, J.; Yin, Y.; Cai, X.; Wang, K.; Wang, G.; Ba, Y.; et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 2009, 28, 1385–1392. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yu, X.; Guo, X.; Tian, Z.; Su, M.; Long, Y.; Huang, C.; Zhou, F.; Liu, M.; Wu, X. miR-143 is downregulated in cervical cancer and promotes apoptosis and inhibits tumor formation by targeting Bcl-2. Mol. Med. Rep. 2012, 5, 753–760. [Google Scholar] [PubMed]
- Clapé, C.; Fritz, V.; Henriquet, C.; Apparailly, F.; Fernandez, P.L.; Iborra, F.; Avancès, C.; Villalba, M.; Culine, S.; Fajas, L. miR-143 interferes with ERK5 signaling, and abrogates prostate cancer progression in mice. PLoS ONE 2009, 4, e7542. [Google Scholar] [CrossRef] [Green Version]
- Meerson, A.; Traurig, M.; Ossowski, V.; Fleming, J.; Mullins, M.; Baier, L. Human adipose microRNA-221 is upregulated in obesity and affects fat metabolism downstream of leptin and TNF-α. Diabetologia 2013, 56, 1971–1979. [Google Scholar] [CrossRef] [Green Version]
- Hwang, M.S.; Yu, N.; Stinson, S.Y.; Yue, P.; Newman, R.J.; Allan, B.B.; Dornan, D. miR-221/222 targets adiponectin receptor 1 to promote the epithelial-to-mesenchymal transition in breast cancer. PLoS ONE 2013, 8, e66502. [Google Scholar]
- Shu, X.; Hildebrandt, M.A.; Gu, J.; Tannir, N.M.; Matin, S.F.; Karam, J.A.; Wood, C.G.; Wu, X. MicroRNA profiling in clear cell renal cell carcinoma tissues potentially links tumorigenesis and recurrence with obesity. Br. J. Cancer 2017, 116, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Creighton, C.J.; Morgan, M.; Gunaratne, P.H.; Wheeler, D.A.; Gibbs, R.A.; Gordon Robertson, A.; Chu, A.; Beroukhim, R.; Cibulskis, K.; Signoretti, S.; et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Chung, T.K.; Lau, T.S.; Cheung, T.H.; Yim, S.F.; Lo, K.W.; Siu, N.S.; Chan, L.K.; Yu, M.Y.; Kwong, J.; Doran, G.; et al. Dysregulation of microRNA-204 mediates migration and invasion of endometrial cancer by regulating FOXC1. Int. J. Cancer 2012, 130, 1036–1045. [Google Scholar] [CrossRef]
- Qiu, Y.H.; Wei, Y.P.; Shen, N.J.; Wang, Z.C.; Kan, T.; Yu, W.L.; Yi, B.; Zhang, Y.J. miR-204 inhibits epithelial to mesenchymal transition by targeting slug in intrahepatic cholangiocarcinoma cells. Cell. Physiol. Biochem. 2013, 32, 1331–1341. [Google Scholar] [CrossRef]
- Ying, Z.; Li, Y.; Wu, J.; Zhu, X.; Yang, Y.; Tian, H.; Li, W.; Hu, B.; Cheng, S.Y.; Li, M. Loss of miR-204 expression enhances glioma migration and stem cell-like phenotype. Cancer Res. 2013, 73, 990–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, R.; Lodish, H.; Sun, L. MicroRNAs in adipogenesis and as therapeutic targets for obesity. Expert Opin. Ther. Targets 2011, 15, 623–636. [Google Scholar] [CrossRef]
- Slaby, O.; Redova, M.; Poprach, A.; Nekvindova, J.; Iliev, R.; Radova, L.; Lakomy, R.; Svoboda, M.; Vyzula, R. Identification of MicroRNAs associated with early relapse after nephrectomy in renal cell carcinoma patients. Genes Chromosom. Cancer 2012, 51, 707–716. [Google Scholar] [CrossRef]
- Wu, X.; Weng, L.; Li, X.; Guo, C.; Pal, S.K.; Jin, J.M.; Li, Y.; Nelson, R.A.; Mu, B.; Onami, S.H.; et al. Identification of a 4-microRNA signature for clear cell renal cell carcinoma metastasis and prognosis. PLoS ONE 2012, 7, e35661. [Google Scholar] [CrossRef]
- Elmore, L.W.; Greer, S.F.; Daniels, E.C.; Saxe, C.C.; Melner, M.H.; Krawiec, G.M.; Cance, W.G.; Phelps, W.C. Blueprint for cancer research: Critical gaps and opportunities. CA Cancer J. Clin. 2021, 71, 107–139. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gluba-Brzózka, A.; Rysz, J.; Ławiński, J.; Franczyk, B. Renal Cell Cancer and Obesity. Int. J. Mol. Sci. 2022, 23, 3404. https://doi.org/10.3390/ijms23063404
Gluba-Brzózka A, Rysz J, Ławiński J, Franczyk B. Renal Cell Cancer and Obesity. International Journal of Molecular Sciences. 2022; 23(6):3404. https://doi.org/10.3390/ijms23063404
Chicago/Turabian StyleGluba-Brzózka, Anna, Jacek Rysz, Janusz Ławiński, and Beata Franczyk. 2022. "Renal Cell Cancer and Obesity" International Journal of Molecular Sciences 23, no. 6: 3404. https://doi.org/10.3390/ijms23063404
APA StyleGluba-Brzózka, A., Rysz, J., Ławiński, J., & Franczyk, B. (2022). Renal Cell Cancer and Obesity. International Journal of Molecular Sciences, 23(6), 3404. https://doi.org/10.3390/ijms23063404