
Autophagy Alteration in ApoA-I Related Systemic Amyloidosis

 ,
,  ,
,  ,
, 
Abstract
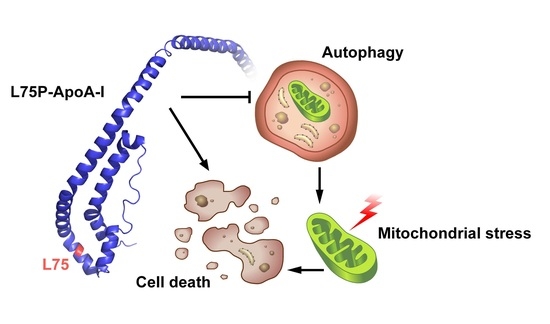
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
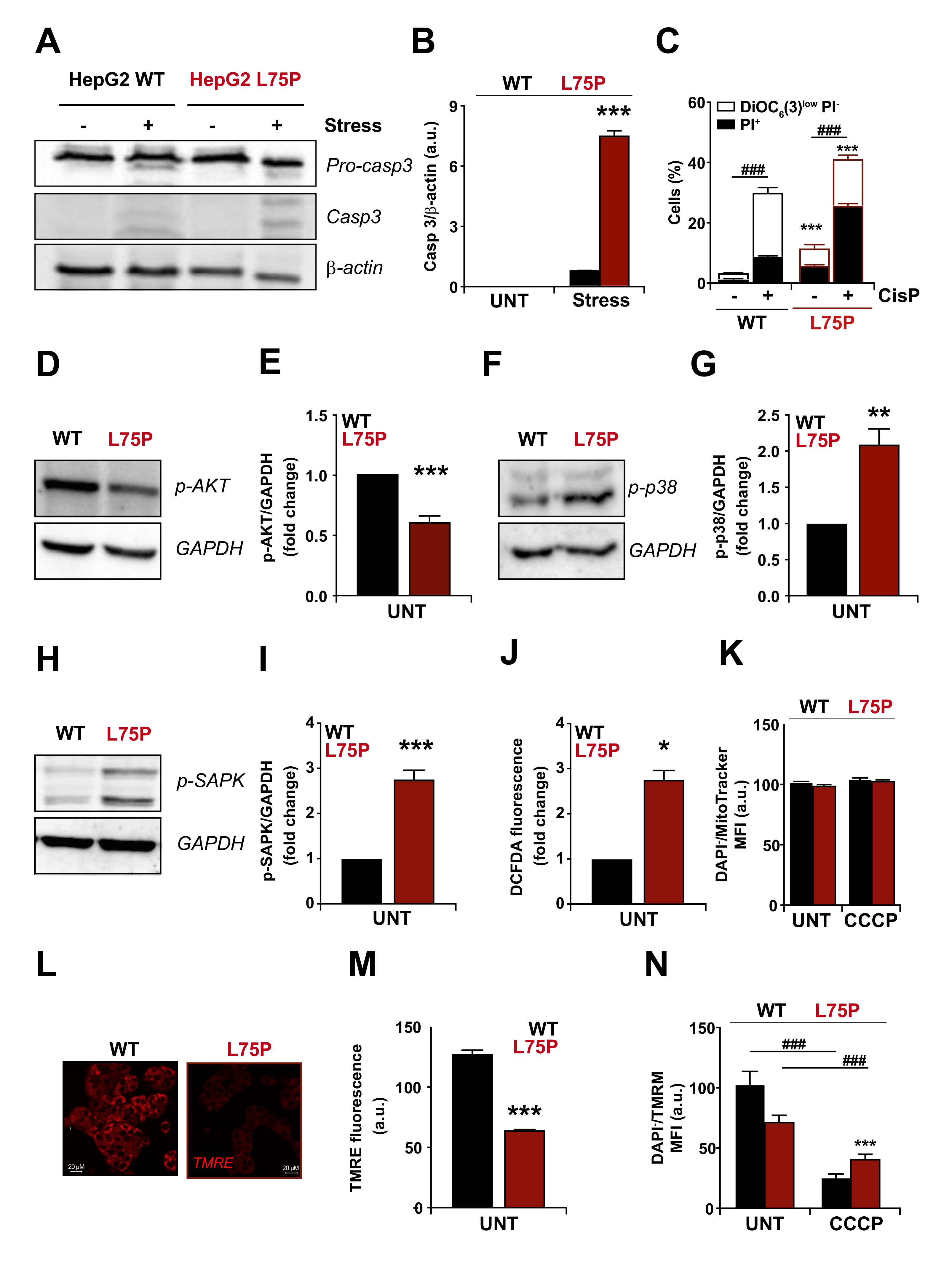
2.1. L75P-ApoA-I Variant Compromises Cellular Redox Homeostasis and Causes Mitochondrial Dysfunction
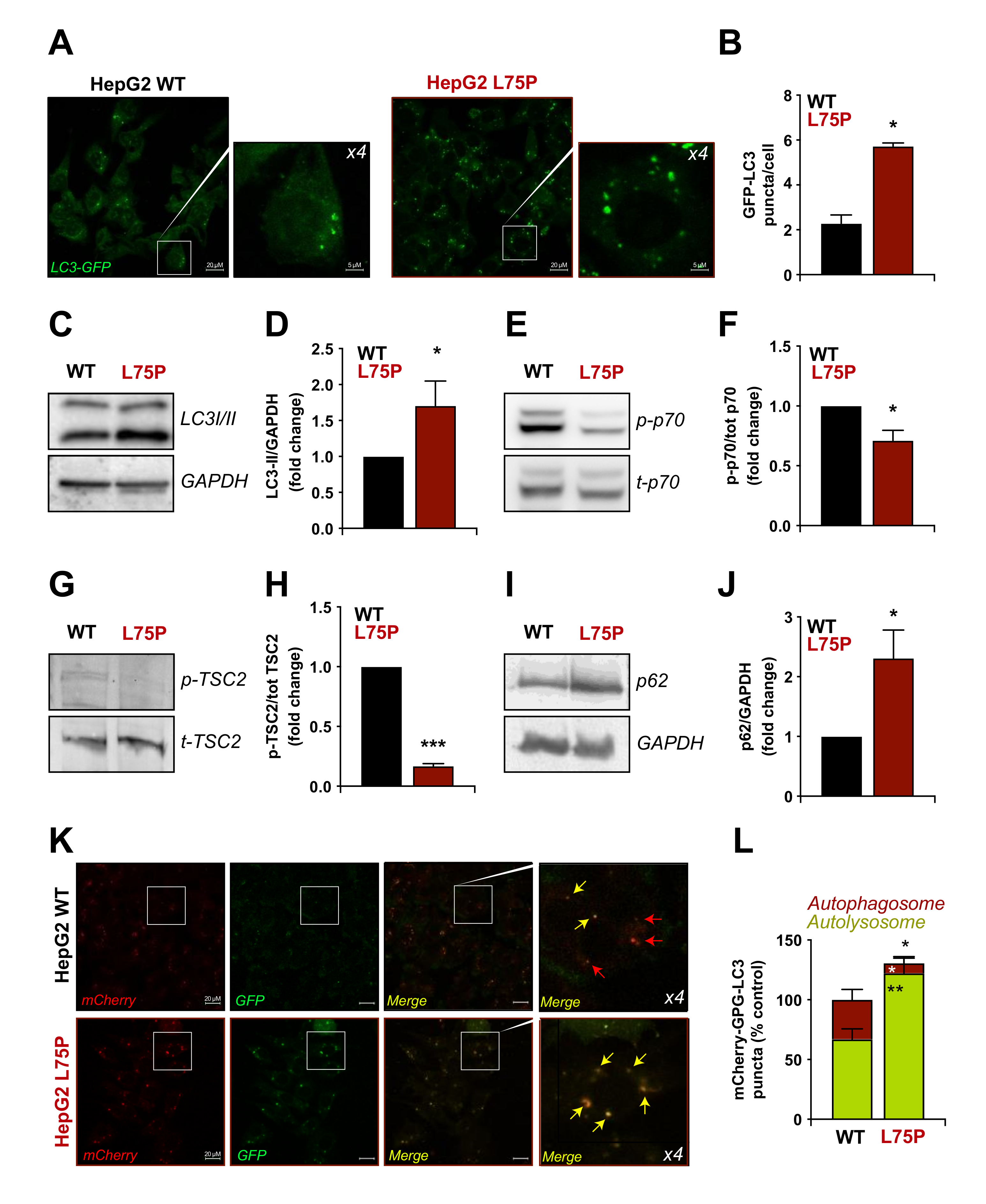
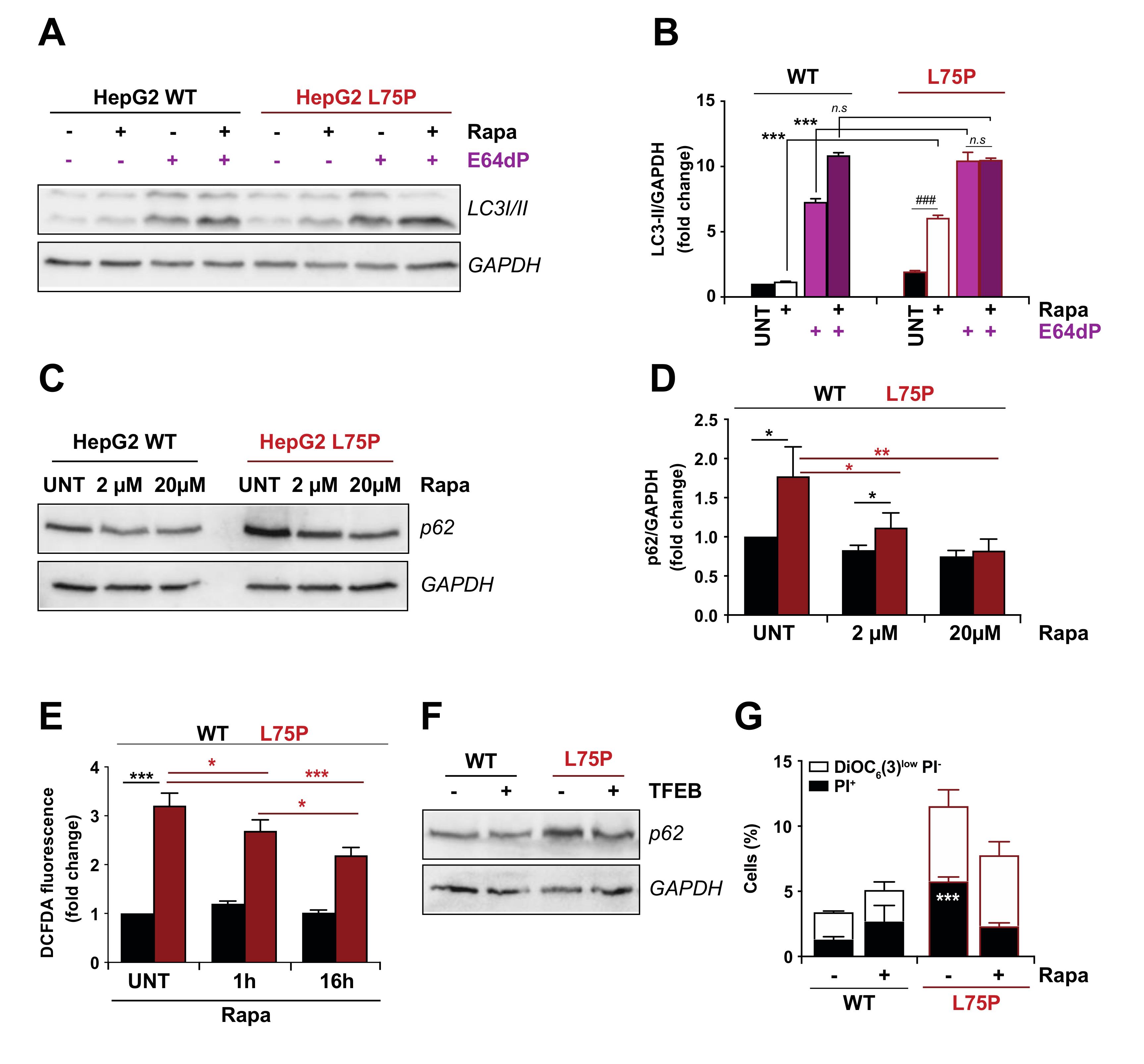
2.2. L75P-ApoA-I Induces an Impairment of the Autophagic Flux
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Culture Conditions and Treatments
4.2. Plasmids Transfection
4.3. Western Blot
4.4. DCFDA Assay
4.5. TMRE Assay
4.6. Cytofluorometric Assessment of Apoptosis
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | protein kinase B |
| AL | light chain |
| ApoA-I | apolipoprotein A1 |
| Atg | autophagy-related |
| BSA | bovine serum albumin |
| DMEM | Dulbecco’s Modification of Eagle’s Medium |
| FBS | foetal bovine serum |
| G418 | geneticin |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GFP | green fluorescent protein |
| H2-DCFDA | 2′,7′-dichlorodihydrofluorescein diacetate |
| HepG2 | human hepatocellular carcinoma |
| LC3 | microtubule associated protein 1 light chain 3 (MAP1LC3) |
| Leu | leucine |
| MAPK | mitogen-activated protein kinase |
| MTOR | mammalian target of Rapamycin |
| p70 | ribosomal protein S6 kinase beta 1 (RPS6KB1, p70S6K) |
| PE | phosphatidylethanolamine |
| Pep-A | pepstatin-A |
| Pro | proline |
| ROS | reactive oxygen species |
| SAPK/JNK | stress-activated protein kinase/Jun-amino-terminal kinase |
| SD | standard deviation |
| SEM | standard error of the mean |
| SQSTM1/p62 | sequestosome 1 |
| TFEB | transcription factor EB |
| TMRE | tetramethylrhodamine ethyl ester |
| TSC2 | tuberous sclerosis 2 |
| UV | ultraviolet |
| WT | wild-type |
| Δψm | mitochondrial membrane potential |
References
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B. Amyloidosis. Annu. Rev. Med. 2006, 57, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Schönland, S.; Yumlu, S.; Hegenbart, U.; von Hutten, H.; Gioeva, Z.; Lohse, P.; Büttner, J.; Schmidt, H.; Röcken, C. Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens: Identification of three novel mutations in the APOA1 gene. J. Mol. Diagn. 2009, 11, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Vonberg, F.W.; Gilbertson, J.A.; Rowczenio, D.; Hutt, D.F.; Rendell, N.; Taylor, G.; Hawkins, P.N.; Gillmore, J.D. Amyloid cardiomyopathy associated with a novel apolipoprotein A-I Q172P variant. Amyloid 2015, 22, 252–253. [Google Scholar] [CrossRef]
- Obici, L.; Franceschini, G.; Calabresi, L.; Giorgetti, S.; Stoppini, M.; Merlini, G.; Bellotti, V. Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid 2006, 13, 191–205. [Google Scholar] [CrossRef]
- Rowczenio, D.; Dogan, A.; Theis, J.D.; Vrana, J.A.; Lachmann, H.J.; Wechalekar, A.D.; Gilbertson, J.A.; Hunt, T.; Gibbs, S.D.J.; Sattianayagam, P.T.; et al. Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I. Am. J. Pathol. 2011, 179, 1978–1987. [Google Scholar] [CrossRef]
- Horike, K.; Takeda, A.; Tsujita, M.; Goto, N.; Watarai, Y.; Uchida, K.; Katayama, A.; Nishihira, M.; Shimizu, A.; Nozu, K.; et al. Two novel APOA1 gene mutations in a Japanese renal transplant recipient with recurrent apolipoprotein A-I related amyloidosis. Nephrology 2018, 23, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Morgado, I.; Panahi, A.; Burwash, A.G.; Das, M.; Straub, J.E.; Gursky, O. Molecular Insights into Human Hereditary Apolipoprotein A-I Amyloidosis Caused by the Glu34Lys Mutation. Biochemistry 2018, 57, 5738–5747. [Google Scholar] [CrossRef]
- Arciello, A.; Piccoli, R.; Monti, D.M. Apolipoprotein A-I: The dual face of a protein. FEBS Lett. 2016, 590, 4171–4179. [Google Scholar] [CrossRef] [Green Version]
- Del Giudice, R.; Arciello, A.; Itri, F.; Merlino, A.; Monti, M.; Buonanno, M.; Penco, A.; Canetti, D.; Petruk, G.; Monti, S.M.; et al. Protein conformational perturbations in hereditary amyloidosis: Differential impact of single point mutations in ApoAI amyloidogenic variants. Biochim. Biophys. Acta 2016, 1860, 434–444. [Google Scholar] [CrossRef]
- Raimondi, S.; Guglielmi, F.; Giorgetti, S.; Di Gaetano, S.; Arciello, A.; Monti, D.M.; Relini, A.; Nichino, D.; Doglia, S.M.; Natalello, A.; et al. Effects of the known pathogenic mutations on the aggregation pathway of the amyloidogenic peptide of apolipoprotein A-I. J. Mol. Biol. 2011, 407, 465–476. [Google Scholar] [CrossRef]
- Mangione, P.; Sunde, M.; Giorgetti, S.; Stoppini, M.; Esposito, G.; Gianelli, L.; Obici, L.; Asti, L.; Andreola, A.; Viglino, P.; et al. Amyloid fibrils derived from the apolipoprotein A1 Leu174Ser variant contain elements of ordered helical structure. Protein Sci. 2001, 10, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Del Giudice, R.; Domingo-Espín, J.; Iacobucci, I.; Nilsson, O.; Monti, M.; Monti, D.M.; Lagerstedt, J.O. Structural determinants in ApoA-I amyloidogenic variants explain improved cholesterol metabolism despite low HDL levels. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3038–3048. [Google Scholar] [CrossRef]
- Das, M.; Wilson, C.J.; Mei, X.; Wales, T.E.; Engen, J.R.; Gursky, O. Structural Stability and Local Dynamics in Disease-Causing Mutants of Human Apolipoprotein A-I: What Makes the Protein Amyloidogenic? J. Mol. Biol. 2016, 428, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Lagerstedt, J.O.; Cavigiolio, G.; Roberts, L.M.; Hong, H.S.; Jin, L.W.; Fitzgerald, P.G.; Oda, M.N.; Voss, J.C. Mapping the structural transition in an amyloidogenic apolipoprotein A-I. Biochemistry 2007, 46, 9693–9699. [Google Scholar] [CrossRef] [Green Version]
- Petrlova, J.; Duong, T.; Cochran, M.C.; Axelsson, A.; Mörgelin, M.; Roberts, L.M.; Lagerstedt, J.O. The fibrillogenic L178H variant of apolipoprotein A-I forms helical fibrils. J. Lipid Res. 2012, 53, 390–398. [Google Scholar] [CrossRef] [Green Version]
- Obici, L.; Palladini, G.; Giorgetti, S.; Bellotti, V.; Gregorini, G.; Arbustini, E.; Verga, L.; Marciano, S.; Donadei, S.; Perfetti, V.; et al. Liver biopsy discloses a new apolipoprotein A-I hereditary amyloidosis in several unrelated Italian families. Gastroenterology 2004, 126, 1416–1422. [Google Scholar] [CrossRef]
- Del Giudice, R.; Monti, D.M.; Sarcinelli, C.; Arciello, A.; Piccoli, R.; Hu, G.F. Amyloidogenic variant of apolipoprotein A-I elicits cellular stress by attenuating the protective activity of angiogenin. Cell Death Dis. 2014, 5, e1097. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otin, C.; Kroemer, G. Hallmarks of health. Cell 2021, 184, 1929–1939. [Google Scholar] [CrossRef]
- Rao, X.; Wang, Y. Apolipoprotein A-I improves hepatic autophagy through the AMPK pathway. Biochimie 2019, 165, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Georgila, K.; Gounis, M.; Havaki, S.; Gorgoulis, V.G.; Eliopoulos, A.G. mTORC1-dependent protein synthesis and autophagy uncouple in the regulation of Apolipoprotein A-I expression. Metabolism 2020, 105, 154186. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 16. [Google Scholar] [CrossRef]
- Georgieva, E.; Ivanova, D.; Zhelev, Z.; Bakalova, R.; Gulubova, M.; Aoki, I. Mitochondrial Dysfunction and Redox Imbalance as a Diagnostic Marker of "Free Radical Diseases". Anticancer Res. 2017, 37, 5373–5381. [Google Scholar]
- Kameyama, H.; Nakajima, H.; Nishitsuji, K.; Mikawa, S.; Uchimura, K.; Kobayashi, N.; Okuhira, K.; Saito, H.; Sakashita, N. Iowa Mutant Apolipoprotein A-I (ApoA-IIowa) Fibrils Target Lysosomes. Sci. Rep. 2016, 6, 30391. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, K.; Nishitsuji, K.; Uchimura, K.; Hung, S.C.; Mizuguchi, M.; Nakajima, H.; Mikawa, S.; Kobayashi, N.; Saito, H.; Sakashita, N. Cellular interaction and cytotoxicity of the iowa mutation of apolipoprotein A-I (ApoA-IIowa) amyloid mediated by sulfate moieties of heparan sulfate. J. Biol. Chem. 2015, 290, 24210–24221. [Google Scholar] [CrossRef] [Green Version]
- Fujikake, N.; Shin, M.; Shimizu, S. Association Between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 225. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Mishra, S.; Qiu, Y.; Shi, J.; Trudeau, K.; Las, G.; Liesa, M.; Shirihai, O.S.; Connors, L.H.; Seldin, D.C.; et al. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO Mol. Med. 2014, 6, 1493–1507. [Google Scholar] [CrossRef]
- Oliva, L.; Orfanelli, U.; Resnati, M.; Raimondi, A.; Orsi, A.; Milan, E.; Palladini, G.; Milani, P.; Cerruti, F.; Cascio, P.; et al. The amyloidogenic light chain is a stressor that sensitizes plasma cells to proteasome inhibitor toxicity. Blood 2017, 129, 2132–2142. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy 3rd edition. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses MTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Bravo-San Pedro, J.M.; Niso-Santano, M.; Gómez-Sánchez, R.; Pizarro-Estrella, E.; Aiastui-Pujana, A.; Gorostidi, A.; Climent, V.; López de Maturana, R.; Sanchez-Pernaute, R.; López de Munain, A.; et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell. Mol. Life Sci. 2013, 70, 121–136. [Google Scholar] [CrossRef]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, T.; Masuda-Suzukake, M.; Hasegawa, M. Molecular mechanisms of the co-deposition of multiple pathological proteins in neurodegenerative diseases. Neuropathology 2018, 38, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, C.J.; La Spada, A.R. TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: Molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol. Dis. 2018, 122, 30150–30155. [Google Scholar] [CrossRef]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The Transcription Factor TFEB Links mTORC1 Signaling to Transcriptional Control of Lysosome Homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Townsend, D.; Hughes, E.; Hussain, R.; Siligardi, G.; Baldock, S.; Madine, J.; Middleton, D.A. Heparin and Methionine Oxidation Promote the Formation of Apolipoprotein A-I Amyloid Comprising α-Helical and β-Sheet Structures. Biochemistry 2017, 56, 1632–1644. [Google Scholar] [CrossRef]
- Jayaraman, S.; Gantz, D.L.; Haupt, C.; Gursky, O. Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis. Proc. Natl. Acad. Sci. USA 2017, 114, E6507–E6515. [Google Scholar] [CrossRef] [Green Version]
- Gaglione, R.; Smaldone, G.; Di Girolamo, R.; Piccoli, R.; Pedone, E.; Arciello, A. Cell milieu significantly affects the fate of AApoAI amyloidogenic variants: Predestination or serendipity? Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 377–384. [Google Scholar] [CrossRef]
- Chisholm, J.W.; Burleson, E.R.; Shelness, G.S.; Parks, J.S. ApoA-I secretion from HepG2 cells: Evidence for the secretion of both lipid-poor apoA-I and intracellularly assembled nascent HDL. J. Lipid Res. 2002, 43, 36–44. [Google Scholar] [CrossRef]
- Nilsson, O.; Lindvall, M.; Obici, L.; Ekström, S.; Lagerstedt, J.O.; Del Giudice, R. Structure dynamics of ApoA-I amyloidogenic variants in small HDL increase their ability to mediate cholesterol efflux. J. Lipid Res. 2021, 62, 100004. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Del Giudice, R.; Petruk, G.; Raiola, A.; Barone, A.; Monti, D.M.; Rigano, M.M. Carotenoids in fresh and processed tomato (Solanum lycopersicum) fruits protect cells from oxidative stress injury. J. Sci. Food Agric. 2017, 97, 1616–1623. [Google Scholar] [CrossRef]
- Monti, D.M.; Ferraro, G.; Petruk, G.; Maiore, L.; Pane, F.; Amoresano, A.; Cinellu, M.A.; Merlino, A. Ferritin nanocages loaded with gold ions induce oxidative stress and apoptosis in MCF-7 human breast cancer cells. Dalton Trans. 2017, 46, 15354–15362. [Google Scholar] [CrossRef] [Green Version]
- Sica, V.; Bravo-San Pedro, J.M.; Izzo, V.; Pol, J.; Pierredon, S.; Enot, D.; Durand, S.; Bossut, N.; Chery, A.; Souquere, S.; et al. Lethal Poisoning of Cancer Cells by Respiratory Chain Inhibition plus Dimethyl α-Ketoglutarate. Cell Rep. 2019, 27, 820–834.e9. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Giudice, R.; Imbimbo, P.; Pietrocola, F.; Martins, I.; De Palma, F.D.E.; Bravo-San Pedro, J.M.; Kroemer, G.; Maiuri, M.C.; Monti, D.M. Autophagy Alteration in ApoA-I Related Systemic Amyloidosis. Int. J. Mol. Sci. 2022, 23, 3498. https://doi.org/10.3390/ijms23073498
Del Giudice R, Imbimbo P, Pietrocola F, Martins I, De Palma FDE, Bravo-San Pedro JM, Kroemer G, Maiuri MC, Monti DM. Autophagy Alteration in ApoA-I Related Systemic Amyloidosis. International Journal of Molecular Sciences. 2022; 23(7):3498. https://doi.org/10.3390/ijms23073498
Chicago/Turabian StyleDel Giudice, Rita, Paola Imbimbo, Federico Pietrocola, Isabelle Martins, Fatima Domenica Elisa De Palma, José Manuel Bravo-San Pedro, Guido Kroemer, Maria Chiara Maiuri, and Daria Maria Monti. 2022. "Autophagy Alteration in ApoA-I Related Systemic Amyloidosis" International Journal of Molecular Sciences 23, no. 7: 3498. https://doi.org/10.3390/ijms23073498
APA StyleDel Giudice, R., Imbimbo, P., Pietrocola, F., Martins, I., De Palma, F. D. E., Bravo-San Pedro, J. M., Kroemer, G., Maiuri, M. C., & Monti, D. M. (2022). Autophagy Alteration in ApoA-I Related Systemic Amyloidosis. International Journal of Molecular Sciences, 23(7), 3498. https://doi.org/10.3390/ijms23073498