
Molecular Dynamics Studies on the Structural Stability Prediction of SARS-CoV-2 Variants Including Multiple Mutants

 ,
,
Abstract
:1. Introduction
2. Results
2.1. SARS-CoV-2 S Protein Genome/Protein Analysis in Patients with COVID-19
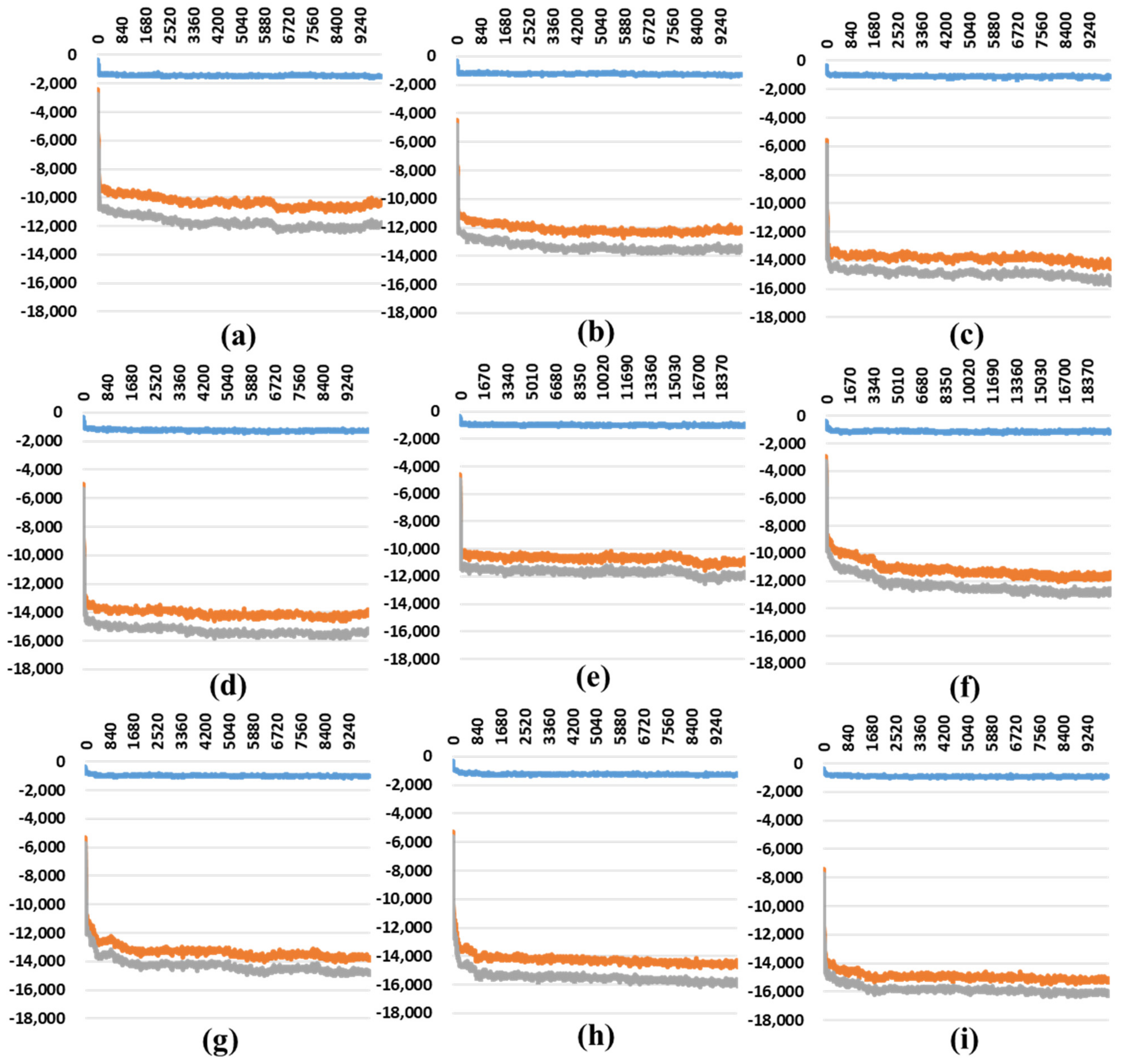
2.2. The Stability Analysis for RBDs in Full-Length Trimeric S Protein Chains
2.3. Binding Free Energy Analysis on Stability between the S Protein and ACE2
3. Discussion
4. Materials and Methods
4.1. Ethical Considerations
4.2. Genome/Protein Sequence Investigation of Korean Patients with COVID-19
4.3. Dataset Preparation for SARS-CoV-2 S Protein Structure
4.4. Self-Homology Modeling for Deletion Mutant
4.5. MD Simulation
4.6. V503 and N501 Residue Distance Calculation
4.7. Binding Free Energy Analysis Using MM/PBSA Calculation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, Y.; Yang, C.; Xu, X.-f.; Xu, W.; Liu, S.-w. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Tiwari, S.; Deb, M.K.; Marty, J.L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): A global pandemic and treatment strategies. Int. J. Antimicrob. Agents 2020, 56, 106054. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, B.; Deepthi, B. Complexity of COVID-19 Dynamics. Entropy 2022, 24, 50. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Zheng, Q.; Zhang, H.; Niu, Y.; Lou, Y.; Wang, H. The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: Implications for the design of spike-based vaccine immunogens. Front. Immunol. 2020, 11, 2593. [Google Scholar] [CrossRef]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Wu, Y.; Zhao, S. Furin cleavage sites naturally occur in coronaviruses. Stem Cell Res. 2021, 50, 102115. [Google Scholar] [CrossRef]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C. Distribution of ACE2, CD147, CD26, and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy 2020, 75, 2829–2845. [Google Scholar] [CrossRef]
- Ray, D.; Le, L.; Andricioaei, I. Distant residues modulate conformational opening in SARS-CoV-2 spike protein. BioRxiv 2021, 118, 43. [Google Scholar] [CrossRef]
- Rath, S.L.; Kumar, K. Investigation of the effect of temperature on the structure of SARS-CoV-2 Spike Protein by Molecular Dynamics Simulations. Front. Mol. Biosci. 2020, 7, 583523. [Google Scholar] [CrossRef]
- Hozhabri, H.; Piceci Sparascio, F.; Sohrabi, H.; Mousavifar, L.; Roy, R.; Scribano, D.; De Luca, A.; Ambrosi, C.; Sarshar, M. The global emergency of novel coronavirus (SARS-CoV-2): An update of the current status and forecasting. Int. J. Environ. Res. Public Health 2020, 17, 5648. [Google Scholar] [CrossRef]
- Piplani, S.; Singh, P.K.; Winkler, D.A.; Petrovsky, N. In silico comparison of spike protein-ACE2 binding affinities across species; significance for the possible origin of the SARS-CoV-2 virus. arXiv 2020, arXiv:2005.06199. [Google Scholar]
- López-Cortés, G.I.; Palacios-Pérez, M.; Zamudio, G.S.; Veledíaz, H.F.; Ortega, E.; José, M.V. Neutral evolution test of the spike protein of SARS-CoV-2 and its implications in the binding to ACE2. Sci. Rep. 2021, 11, 18847. [Google Scholar] [CrossRef]
- Xie, X.; Han, J.-B.; Ma, G.; Feng, X.-L.; Li, X.; Zou, Q.-C.; Deng, Z.-H.; Zeng, J. Emerging SARS-CoV-2 B. 1.621/Mu variant is prominently resistant to inactivated vaccine-elicited antibodies. Zool. Res. 2021, 42, 789. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Hong, W.; Pan, X.; Lu, G.; Wei, X. SARS-CoV-2 Omicron variant: Characteristics and prevention. MedComm 2021, 2, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Sun, Y.; Zhou, J.; Ye, Q. The Global Epidemic of the SARS-CoV-2 Delta Variant, Key Spike Mutations and Immune Escape. Front. Immunol. 2021, 12, 751778. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-E.; Kim, J.-M.; Rhee, J.; Park, A.K.; Kim, E.-J.; Kang, N.S. Molecular Dynamics Studies on the Structural Characteristics for the Stability Prediction of SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 8714. [Google Scholar] [CrossRef]
- Martí, D.; Alsina, M.; Alemán, C.; Bertran, O.; Turon, P.; Torras, J. Unravelling the molecular interactions between the SARS-CoV-2 RBD spike protein and various specific monoclonal antibodies. Biochimie 2021, 193, 90–102. [Google Scholar] [CrossRef]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Nelson, G.; Buzko, O.; Bassett, A.; Spilman, P.; Niazi, K.; Rabizadeh, S.; Soon-Shiong, P. Millisecond-scale molecular dynamics simulation of spike RBD structure reveals evolutionary adaption of SARS-CoV-2 to stably bind ACE2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Deganutti, G.; Prischi, F.; Reynolds, C.A. Supervised molecular dynamics for exploring the druggability of the SARS-CoV-2 spike protein. J. Comput. Aided Mol. Des. 2021, 35, 195–207. [Google Scholar] [CrossRef]
- Peng, C.; Zhu, Z.; Shi, Y.; Wang, X.; Mu, K.; Yang, Y.; Zhang, X.; Xu, Z. Exploring the binding mechanism and accessible angle of SARS-CoV-2 spike and ACE2 by molecular dynamics simulation and free energy calculation. ChemRxiv 2021. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e819. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B. 1.1. 7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef] [PubMed]
- Hamill, V.; Noll, L.; Lu, N.; Tsui, W.N.T.; Porter, E.P.; Gray, M.; Sebhatu, T.; Goerl, K.; Brown, S.; Palinski, R. Molecular detection of SARS-CoV-2 strains and differentiation of Delta variant strains. Transbound. Emerg. Dis. 2021. ahead of print. [Google Scholar] [CrossRef]
- Miner, J.C.; Lappala, A.; Fenimore, P.W.; Fischer, W.M.; McMahon, B.H.; Hengartner, N.W.; Sanbonmatsu, K.Y.; Tung, C.-S. Modeling the Influenza A NP-vRNA-Polymerase Complex in Atomic Detail. Biomolecules 2021, 11, 124. [Google Scholar] [CrossRef]
- Krumm, Z.A.; Lloyd, G.M.; Francis, C.P.; Nasif, L.H.; Mitchell, D.A.; Golde, T.E.; Giasson, B.I.; Xia, Y. Precision therapeutic targets for COVID-19. Virol. J. 2021, 18, 1–22. [Google Scholar] [CrossRef]
- Giron, C.C.; Laaksonen, A.; Barroso da Silva, F.L. Up state of the SARS-CoV-2 spike homotrimer favors an increased virulence for new variants. Front. Med. Technol. 2021, 3, 694347. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, S.; Wu, B.; Yang, Q.; Chen, A.; Li, Y.; Zhang, Y.; Pan, T.; Zhang, H.; He, X. SARS-CoV-2 Omicron strain exhibits potent capabilities for immune evasion and viral entrance. Signal Transduct. Target. Ther. 2021, 6, 1–3. [Google Scholar] [CrossRef]
- Koleya, T.; Kumara, M.; Goswami, A.; Ethayathulla, A.S.; Hariprasada, G. Structural modeling of Omicron spike protein and its complex with human ACE-2 receptor: Molecular basis for high transmissibility of the virus. Biochem. Biophys. Res. Commun. 2022, 592, 51–53. [Google Scholar] [CrossRef]
- Socher, E.; Heger, L.; Paulsen, F.; Zunke, F.; Arnold, P. Molecular dynamics simulations of the delta and omicron SARS-CoV-2 spike–ACE2 complexes reveal distinct changes between both variants. Comput. Struct. Biotechnol. J. 2022. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Chung, Y.-S.; Jo, H.J.; Lee, N.-J.; Kim, M.S.; Woo, S.H.; Park, S.; Kim, J.W.; Kim, H.M.; Han, M.-G. Identification of coronavirus isolated from a patient in Korea with COVID-19. Osong Public Health Res. Perspect. 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, A.K.; Kim, I.-H.; Kim, J.; Kim, J.-M.; Kim, H.M.; Young Lee, C.; Han, M.-G.; Rhie, G.-E.; Kwon, D.; Nam, J.-G. Genomic surveillance of SARS-CoV-2: Distribution of clades in the Republic of Korea in 2020. Osong Public Health Res. Perspect. 2021, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Socher, E.; Conrad, M.; Heger, L.; Paulsen, F.; Sticht, H.; Zunke, F.; Arnold, P. Mutations in the B. 1.1. 7 SARS-CoV-2 spike protein reduce receptor-binding affinity and induce a flexible link to the fusion peptide. Biomedicines 2021, 9, 525. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM force field in GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef]






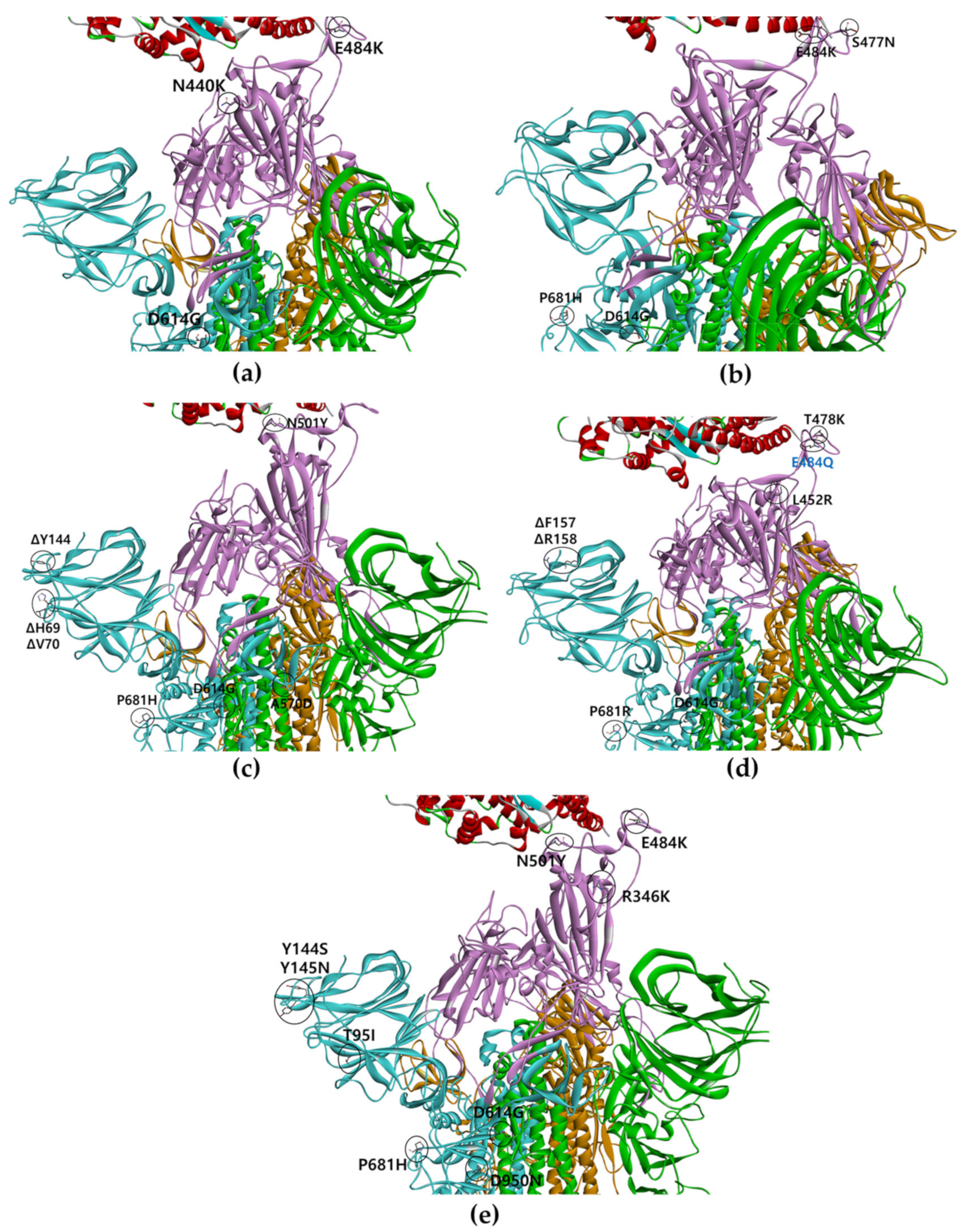{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
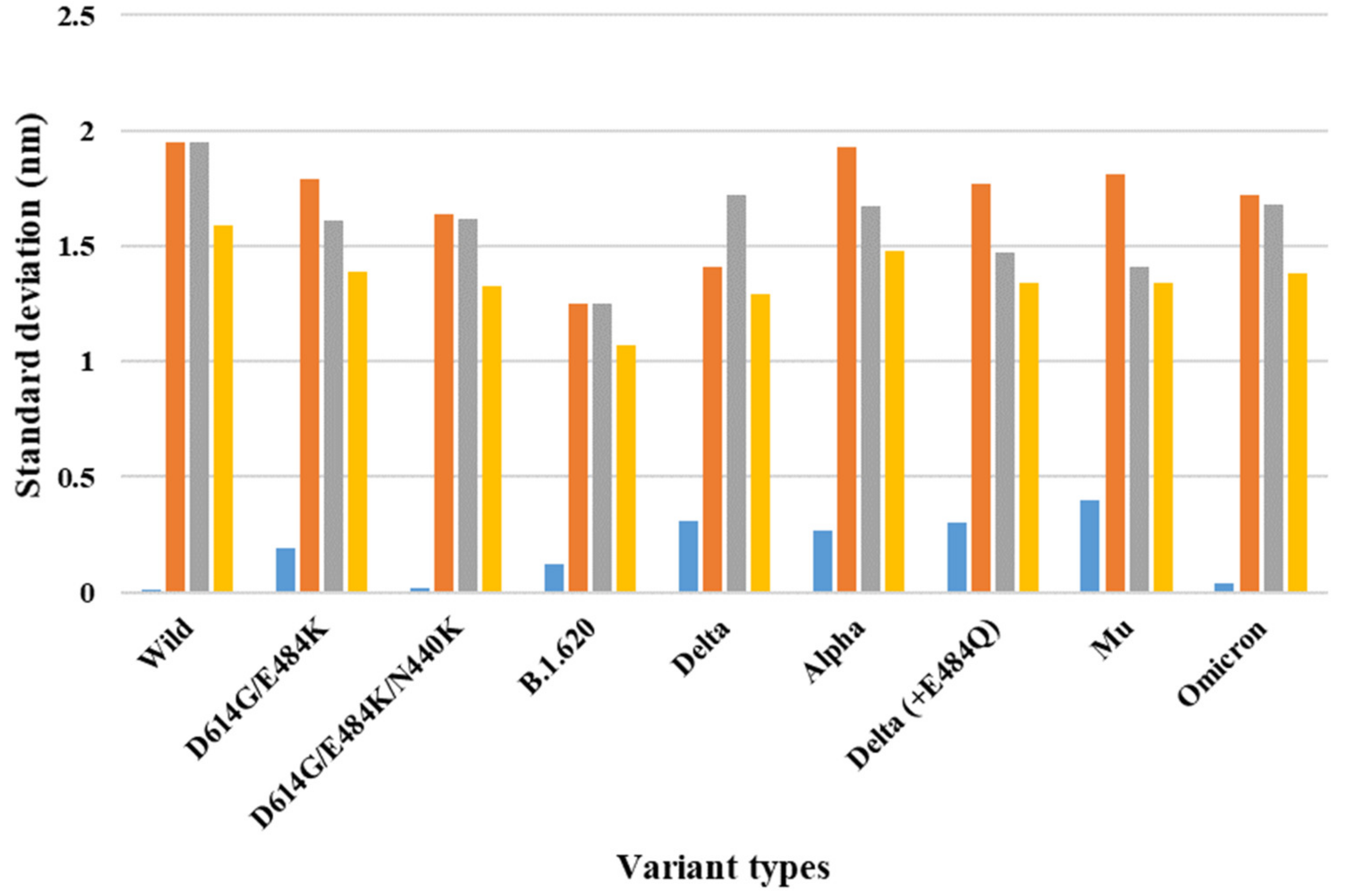
| PDB | Variant Type | A–B | A–C | B–C | S.D. (A–B) & (A–C) | S.D. (A–B) & (B–C) | S.D. (A–C) & (B–C) | S.D. (A–B–C) |
|---|---|---|---|---|---|---|---|---|
| 7A94 (one-open-complex form) | Wild | 3.00 | 3.01 | 0.25 | 0.01 | 1.95 | 1.95 | 1.59 |
| D614G/E484K | 2.78 | 2.51 | 0.24 | 0.19 | 1.79 | 1.61 | 1.39 | |
| D614G/E484K/N440K | 2.78 | 2.75 | 0.46 | 0.02 | 1.64 | 1.62 | 1.33 | |
| B.1.620 | 2.55 | 2.72 | 0.78 | 0.12 | 1.25 | 1.25 | 1.07 | |
| Delta | 2.39 | 2.83 | 0.40 | 0.31 | 1.41 | 1.72 | 1.29 | |
| Alpha | 2.96 | 2.58 | 0.22 | 0.27 | 1.93 | 1.67 | 1.48 | |
| Delta (+E484Q) | 3.08 | 2.66 | 0.58 | 0.30 | 1.77 | 1.47 | 1.34 | |
| Mu | 2.95 | 2.39 | 0.40 | 0.40 | 1.81 | 1.41 | 1.34 | |
| Omicron | 3.33 | 3.28 | 0.91 | 0.04 | 1.72 | 1.68 | 1.38 | |
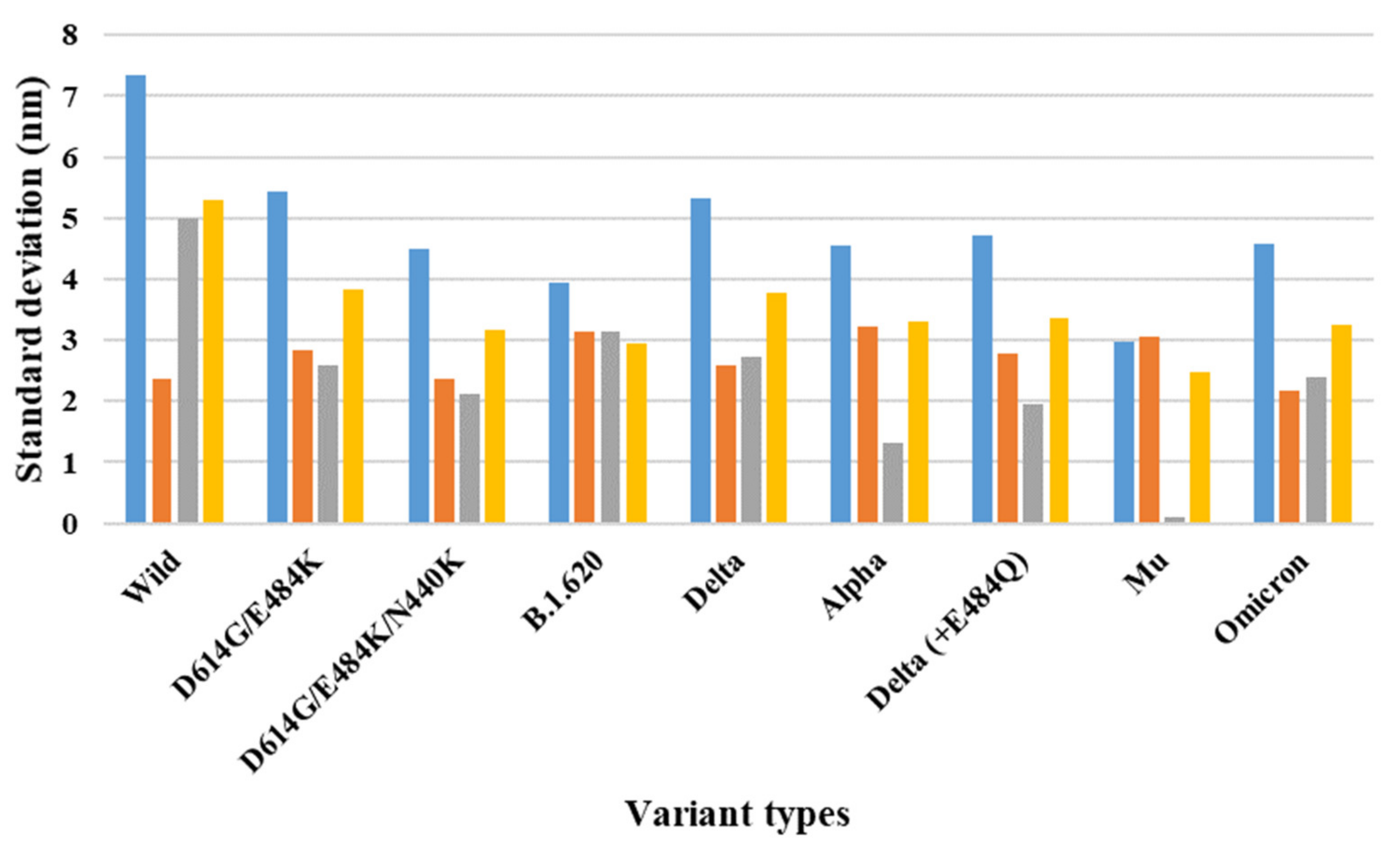
| 7A97 (two-open-complex form) | Wild | 13.96 | 3.57 | 10.63 | 7.35 | 2.36 | 5.00 | 5.31 |
| D614G/E484K | 11.24 | 3.57 | 7.23 | 5.43 | 2.83 | 2.59 | 3.84 | |
| D614G/E484K/N440K | 9.52 | 3.16 | 6.16 | 4.49 | 2.37 | 2.12 | 3.18 | |
| B.1.620 | 9.79 | 4.23 | 5.35 | 3.93 | 3.14 | 3.14 | 2.94 | |
| Delta | 11.13 | 3.59 | 7.45 | 5.33 | 2.60 | 2.73 | 3.77 | |
| Alpha | 10.65 | 4.23 | 6.09 | 4.55 | 3.23 | 1.32 | 3.31 | |
| Delta (+E484Q) | 10.59 | 3.92 | 6.66 | 4.72 | 2.78 | 1.94 | 3.35 | |
| Mu | 8.20 | 4.00 | 3.86 | 2.97 | 3.07 | 0.10 | 2.47 | |
| Omicron | 9.81 | 3.34 | 6.73 | 4.57 | 2.18 | 2.40 | 3.24 | |
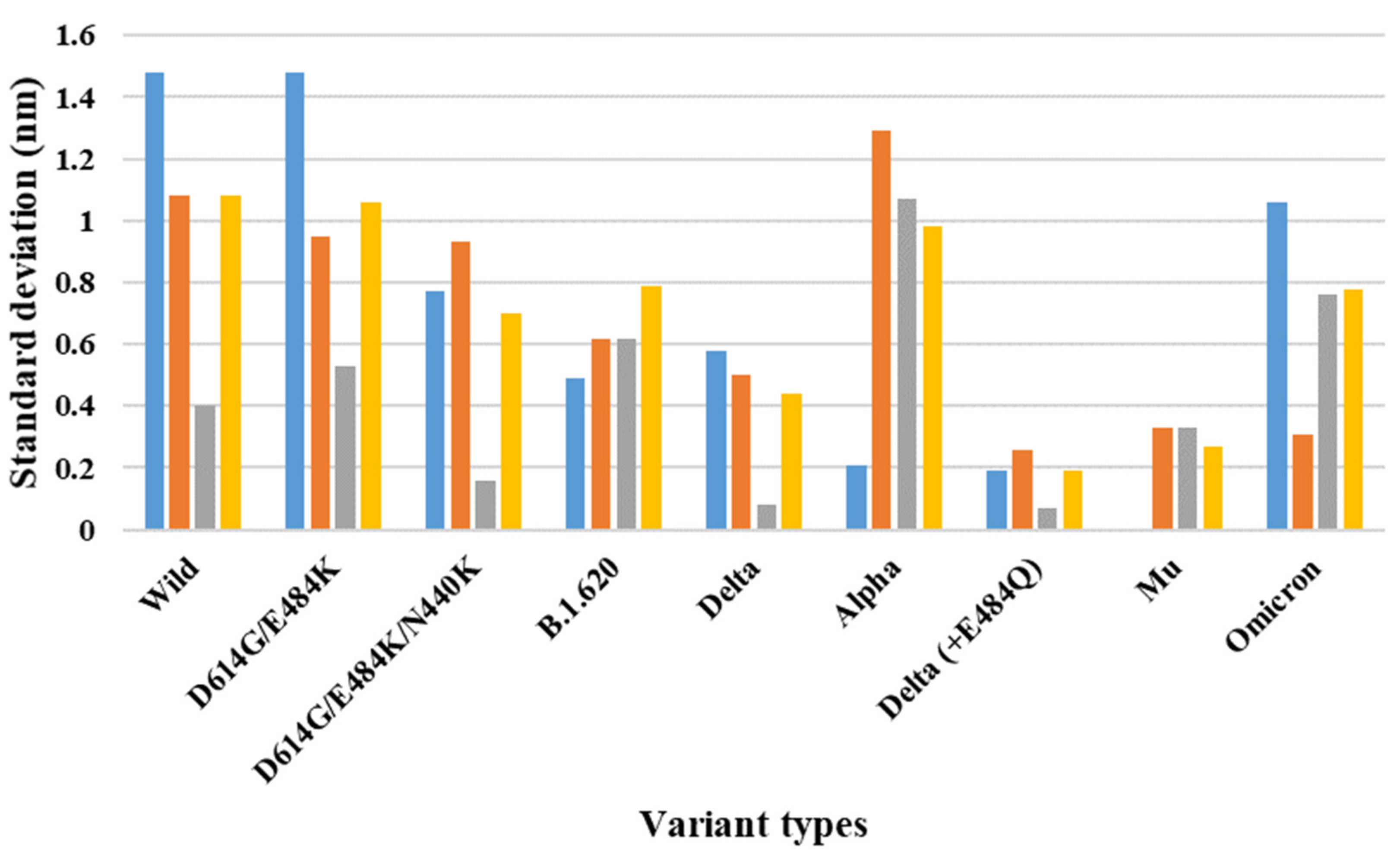
| 7A98 (three-open-complex form) | Wild | 8.23 | 10.32 | 9.75 | 1.48 | 1.08 | 0.40 | 1.08 |
| D614G/E484K | 7.72 | 9.81 | 9.06 | 1.48 | 0.95 | 0.53 | 1.06 | |
| D614G/E484K/N440K | 7.63 | 8.72 | 8.95 | 0.77 | 0.93 | 0.16 | 0.70 | |
| B.1.620 | 8.71 | 8.02 | 9.59 | 0.49 | 0.62 | 0.62 | 0.79 | |
| Delta | 9.18 | 10.00 | 9.88 | 0.58 | 0.50 | 0.08 | 0.44 | |
| Alpha | 8.80 | 9.10 | 10.62 | 0.21 | 1.29 | 1.07 | 0.98 | |
| Delta (+E484Q) | 9.26 | 8.99 | 8.89 | 0.19 | 0.26 | 0.07 | 0.19 | |
| Mu | 9.60 | 9.60 | 9.14 | 0.00 | 0.33 | 0.33 | 0.27 | |
| Omicron | 10.32 | 8.81 | 9.89 | 1.06 | 0.31 | 0.76 | 0.78 |
| Variant Type | V | E | T |
|---|---|---|---|
| Wild | −1461.5 | −10,311.8 | −11,773.3 |
| Wild_deltascale (ΔF157/ΔR158) | −1456.7 | −9227.7 | −10,684.4 |
| Wild_ alphascale (ΔH69/ΔV70/ΔY144) | −1457.6 | −10152.2 | −11609.8 |
| D614G/E484K | −1283.4 | −12,360.2 | −13,643.7 |
| D614G/E484K_deltascale (ΔF157/ΔR158) | −1280.2 | −11,380.0 | −12,660.2 |
| D614G/E484K_alphascale (ΔH69/ΔV70/ΔY144) | −1283.1 | −12,359.2 | −13,642.3 |
| D614G/E484K/N440K | −1116.2 | −14,581.0 | −15,697.2 |
| D614G/E484K/N440K deltascale (ΔF157/ΔR158) | −1114.5 | −13,526.4 | −14,640.9 |
| D614G/E484K/N440K_alphascale (ΔH69/ΔV70/ΔY144) | −1115.0 | −14,570.2 | −15,685.2 |
| B.1.620 (S477N/E484K/D614G/P681H) | −1318.0 | −13,832.5 | −15,150.5 |
| B.1.620_deltascale (ΔF157/ΔR158) | −1316.3 | −12,772.9 | −14,089.2 |
| B.1.620_alphascale (ΔH69/ΔV70/ΔY144) | −1317.7 | −13,821.9 | −15,139.6 |
| Delta (ΔF157/ΔR158/L452R/T478K/D614G/P681R) | −968.9 | −11,150.0 | −12,118.9 |
| Alpha (ΔH69/ΔV70/ΔY144/N501Y/A570D/D614G/P681H) | −1157.5 | −11,690.1 | −12,847.7 |
| Delta_E484Q (VF157/ΔR158/L452R/T478K/D614G/P681R/E484Q) | −1046.3 | −13,738.3 | −14,784.6 |
| Mu (T95I/Y144S/Y145N/R346K/E484K/N501Y/D614G/P681H/D950N) | −1290.4 | −14,776.8 | −16,067.2 |
| Mu_deltascale (ΔF157/ΔR158) | −1280.3 | −13,360.1 | −14,640.4 |
| Mu_alphascale (ΔH69/ΔV70/ΔY144) | −1290.0 | −14,782.2 | −16,072.2 |
| Omicron | −921.2 | −16,059.5 | −16,980.7 |
| Variant Type | 7A97 AD | 7A97 BE | 7A97 Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| V | E | T | V | E | T | V | E | T | |
| Wild | −1326.4 | −13,200.4 | −14,526.7 | −677.3 | −5553.0 | −6230.3 | −2003.7 | −18,753.4 | −20,757.0 |
| Wild_deltascale (ΔF157/ΔR158) | −1305.0 | −11,646.9 | −12,951.9 | −677.2 | −5195.1 | −5872.3 | −1982.3 | −16,842.0 | −18,824.3 |
| Wild_alphascale (ΔH69/ΔV70/ΔY144) | −1311.7 | −12,967.2 | −14,278.9 | −677.2 | −5550.9 | −6228.1 | −1989.0 | −18,518.1 | −20,507.1 |
| D614G/E484K | −1265.9 | −13,766.8 | −15,032.8 | −725.2 | −8535.5 | −9260.7 | −2003.2 | −20,445.4 | −22,448.6 |
| D614G/E484K_deltascale (ΔF157/ΔR158) | −1278.0 | −12,305.2 | −13,583.2 | −725.2 | −8140.3 | −8865.5 | −2003.2 | −20,445.6 | −22,448.8 |
| D614G/E484K_alphascale (ΔH69/ΔV70/ΔY144) | −1250.0 | −13,611.7 | −14,861.7 | −725.2 | −8538.6 | −9263.8 | −1975.3 | −22,150.4 | −24,125.6 |
| D614G/E484K/N440K | −874.3 | −13,307.9 | −14,182.2 | −626.7 | −10,773.4 | −11,400.1 | −1504.7 | −22,321.5 | −23,826.2 |
| D614G/E484K/N440K_deltascale (ΔF157/ΔR158) | −878.0 | −11,944.6 | −12,822.6 | −626.7 | −10,376.9 | −11,003.6 | −1504.7 | −22,321.5 | −23,826.2 |
| D614G/E484K/N440K_alphascale (ΔH69/ΔV70/ΔY144) | −873.4 | −13,308.3 | −14,181.8 | −626.7 | −10,774.4 | −11,401.1 | −1500.1 | −24,082.7 | −25,582.9 |
| B.1.620 (S477N/E484K/D614G/P681H) | −1696.4 | −14,874.1 | −16,570.6 | −676.0 | −10,506.1 | −11,182.1 | −2368.8 | −23,361.0 | −25,729.8 |
| B.1.620_deltascale (ΔF157/ΔR158) | −1692.8 | −13,415.8 | −15,108.6 | −676.0 | −9945.1 | −10,621.1 | −2368.8 | −23,360.9 | −25,729.7 |
| B.1.620_alphascale (ΔH69/ΔV70/ΔY144) | −1647.6 | −14,698.5 | −16,346.1 | −676.0 | −10,500.0 | −11,176.0 | −2323.6 | −25,198.5 | −27,522.1 |
| Delta (ΔF157/ΔR158/L452R/T478K/D614G/P681R) | −1344.8 | −13,867.5 | −15,212.3 | −534.5 | −8634.5 | −9169.0 | −1879.3 | −22,502.0 | −24,831.3 |
| Alpha (ΔH69/ΔV70/ΔY144/N501Y/A570D/D614G/P681H) | −1273.8 | −11,310.1 | −12,583.9 | −917.2 | −6604.0 | −7521.2 | −2191.0 | −17,914.1 | −20,105.1 |
| Delta_E484Q (ΔF157/ΔR158/L452R/T478K/D614G/P681R/E484Q) | −1385.1 | −13,658.2 | −15,043.3 | −676.6 | −10,319.8 | −10,996.4 | −2061.7 | −23,978.0 | −26,039.7 |
| Mu (T95I/Y144S/Y145N/R346K/E484K/N501Y/D614G/P681H/D950N) | −1084.8 | −13,097.6 | −14,182.3 | −1356.9 | −14,208.0 | −15,564.9 | −2441.7 | −27,305.6 | −29,747.2 |
| Mu_deltascale (ΔF157/ΔR158) | −1093.6 | −11,809.3 | −12,902.9 | −1356.4 | −13,230.0 | −14,586.4 | −2450.0 | −25,039.3 | −27,489.3 |
| Mu_alphascale (ΔH69/ΔV70/ΔY144) | −1074.1 | −13,035.9 | −14,110.0 | −1355.7 | −14,169.2 | −15,524.9 | −2429.8 | −27,205.1 | −29,634.9 |
| Omicron | −1343.9 | −16,994.6 | −18,338.5 | −1215.5 | −17,733.8 | −18,949.3 | −2559.5 | −34,728.4 | −37,287.8 |
| Variant Type | 7A98 AD | 7A98 BE | 7A98 CF | 7A98 Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V | E | T | V | E | T | V | E | T | V | E | T | |
| Wild | −535.4 | −5278.5 | −5813.8 | −527.7 | −4872.3 | −5400.0 | −583.0 | −5587.4 | −6170.4 | −1646.1 | −15,738.2 | −17,384.2 |
| Wild_deltascale(ΔF157/ΔR158) | −535.4 | −4883.9 | −5419.3 | −527.7 | −4404.4 | −4932.1 | −583.0 | −5176.7 | −5759.7 | −1646.0 | −14,465.0 | −16,111.0 |
| Wild_ alphascale (ΔH69/ΔV70/ΔY144) | −535.4 | −5276.2 | −5811.6 | −527.7 | −4873.3 | −5401.0 | −583.0 | −5586.5 | −6169.5 | −1646.0 | −15,736.1 | −17,382.1 |
| D614G/E484K | −1236.3 | −12,616.4 | −13,852.7 | −931.4 | −13,292.4 | −14,223.9 | −710.6 | −10,229.1 | −10,939.7 | −2878.3 | −36,137.9 | −39,016.3 |
| D614G/E484K_deltascale (ΔF157/ΔR158) | −1236.0 | −11,740.8 | −12,976.9 | −931.1 | −12,398.8 | −13,329.9 | −710.3 | −9402.2 | −10,112.6 | −2877.4 | −33,541.8 | −36,419.4 |
| D614G/E484K_ alphascale (ΔH69/ΔV70/ΔY144) | −1209.9 | −12,601.0 | −13,810.9 | −909.4 | −13,278.9 | −14,188.3 | −717.8 | −10,124.4 | −10,842.2 | −2837.1 | −36,004.3 | −38,841.4 |
| D614G/E484K/N440K | −1203.1 | −12,512.8 | −13,716.0 | −1351.0 | −15,489.1 | −16,840.2 | −1010.8 | −12,006.6 | −13,017.4 | −3565.0 | −40,008.5 | −43,573.6 |
| D614G/E484K/N440K_deltascale (ΔF157/ΔR158) | −1202.8 | −11,658.0 | −12,860.9 | −1350.7 | −14,612.7 | −15,963.4 | −1009.5 | −11,050.2 | −12,059.8 | −3563.0 | −37,320.9 | −40,884.1 |
| D614G/E484K/N440K_alphascale (ΔH69/ΔV70/ΔY144) | −1171.4 | −12,430.8 | −13,602.2 | −1276.9 | −15,413.7 | −16,690.5 | −1000.6 | −11,966.5 | −12,967.1 | −3448.8 | −39,811.0 | −43,259.8 |
| B.1.620 (S477N/E484K/D614G/P681H) | −866.3 | −11,861.2 | −12,727.5 | −733.9 | −10,837.8 | −11,571.8 | −1275.6 | −13,000.2 | −14,275.8 | −2875.8 | −35,699.2 | −38,575.1 |
| B.1.620_deltascale (ΔF157/ΔR158) | −866.0 | −10,973.5 | −11,839.5 | −733.7 | −10,017.6 | −10,751.3 | −1275.4 | −12,173.6 | −13,449.0 | −2875.1 | −33,164.7 | −36,039.8 |
| B.1.620_alphascale (ΔH69/ΔV70/ΔY144) | −864.6 | −11,860.4 | −12,725.0 | −731.7 | −10,830.2 | −11,561.9 | −1237.9 | −12,914.8 | −14,152.7 | −2834.2 | −35,605.4 | −38,439.6 |
| Delta (ΔF157/ΔR158/L452R/T478K/D614G/P681R) | −724.0 | −8939.1 | −9663.1 | −753.4 | −9047.1 | −9800.4 | −687.1 | −8021.5 | −8708.7 | −2164.5 | −26,007.7 | −28,172.2 |
| Alpha(ΔH69/ΔV70/ΔY144/N501Y/A570D/D614G/P681H) | −939.2 | −9293.1 | −10,232.4 | −950.8 | −8220.7 | −9171.4 | −684.4 | −7538.3 | −8222.7 | −2574.4 | −25,052.1 | −27,626.5 |
| Delta_E484Q(ΔF157/ΔR158/L452R/T478K/D614G/P681R/E484Q) | −1017.0 | −12,096.5 | −13,113.4 | −1048.5 | −10,457.3 | −11,505.8 | −1022.2 | −12,888.7 | −13,910.9 | −3087.6 | −35,442.5 | −38,530.1 |
| Mu(T95I/Y144S/Y145N/R346K/E484K/N501Y/D614G/P681H/D950N) | −1168.3 | −12,901.2 | −14,069.5 | −632.7 | −11,080.4 | −11,713.1 | −1258.5 | −13,583.7 | −14,842.2 | −3059.5 | −37,565.3 | −40,624.8 |
| Mu_deltascale (ΔF157/ΔR158) | −1168.1 | −12,064.9 | −13,233.1 | −632.3 | −10,248.1 | −10,880.4 | −1258.0 | −12,631.4 | −13,889.4 | −3058.4 | −34,944.4 | −38,002.9 |
| Mu_alphascale (ΔH69/ΔV70/ΔY144) | −1147.2 | −12,842.0 | −13,989.3 | −630.7 | −11,062.9 | −11,693.6 | −1209.4 | −13,442.6 | −14,652.0 | −2987.3 | −37,347.5 | −40,334.9 |
| Omicron | −872.7 | −14,757.3 | −15,630.0 | −570.9 | −13,743.9 | −14,314.8 | −1081.3 | −14,247.0 | −15,328.3 | −2524.9 | −42,748.2 | −45,273.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, K.-E.; Kim, J.-M.; Rhee, J.E.; Park, A.K.; Kim, E.-J.; Yoo, C.K.; Kang, N.S. Molecular Dynamics Studies on the Structural Stability Prediction of SARS-CoV-2 Variants Including Multiple Mutants. Int. J. Mol. Sci. 2022, 23, 4956. https://doi.org/10.3390/ijms23094956
Choi K-E, Kim J-M, Rhee JE, Park AK, Kim E-J, Yoo CK, Kang NS. Molecular Dynamics Studies on the Structural Stability Prediction of SARS-CoV-2 Variants Including Multiple Mutants. International Journal of Molecular Sciences. 2022; 23(9):4956. https://doi.org/10.3390/ijms23094956
Chicago/Turabian StyleChoi, Kwang-Eun, Jeong-Min Kim, Jee Eun Rhee, Ae Kyung Park, Eun-Jin Kim, Cheon Kwon Yoo, and Nam Sook Kang. 2022. "Molecular Dynamics Studies on the Structural Stability Prediction of SARS-CoV-2 Variants Including Multiple Mutants" International Journal of Molecular Sciences 23, no. 9: 4956. https://doi.org/10.3390/ijms23094956
APA StyleChoi, K. -E., Kim, J. -M., Rhee, J. E., Park, A. K., Kim, E. -J., Yoo, C. K., & Kang, N. S. (2022). Molecular Dynamics Studies on the Structural Stability Prediction of SARS-CoV-2 Variants Including Multiple Mutants. International Journal of Molecular Sciences, 23(9), 4956. https://doi.org/10.3390/ijms23094956