

Renal Oxygen Demand and Nephron Function: Is Glucose a Friend or Foe?

 ,
,  ,
,
Abstract
:1. Introduction
2. Renal Physiology: A Unique Example Based on Efficient Control of Oxygen Consumption

{kind=link}
{kind=link}
| Organ | Total Blood Flow (L/min) | Parenchimal Flow at Rest (mL/min) | Parenchimal Flow Reserve (mL/min) | Proportion of Total Body O2 Consumption at Rest (%) | OMR Ki (kcal/Kg/day) | O2 Extraction (%) | Organ O2 Consumption(%) Na/ATPase Ca ATPase Other | ||
|---|---|---|---|---|---|---|---|---|---|
| Heart | 4/6 | Coronaric 220/260 | Coronaric 450/600 | 11 | 440 | 75 | 1–5 | 15–30 |
|
| Kidney | 1.2 | Glomerular 90/120 | Glomerular 100/140 | 6–7 | 440 | 10–15 | 70 | _ | Gluco- neogenesis |
| Organ | Flow Regulation in Delivering O2 to the Organ | Maximum increase in O2 Consumption (%) | Prominent Regulator of Performance Maintenance | ||||||
| Heart |
Phasic coronary flow:
|
~ 400% *
|
Filling Volume Change (Frank O–Starling E law)
| ||||||
| Kidney |
Inhomogeneous blood flow:
|
Difficult to assess:
|
Filtration Pressure Change (Starling E law)
| ||||||
3. The Source of Glomerular Filtration Pressure and the Generation of the Filtration Fraction
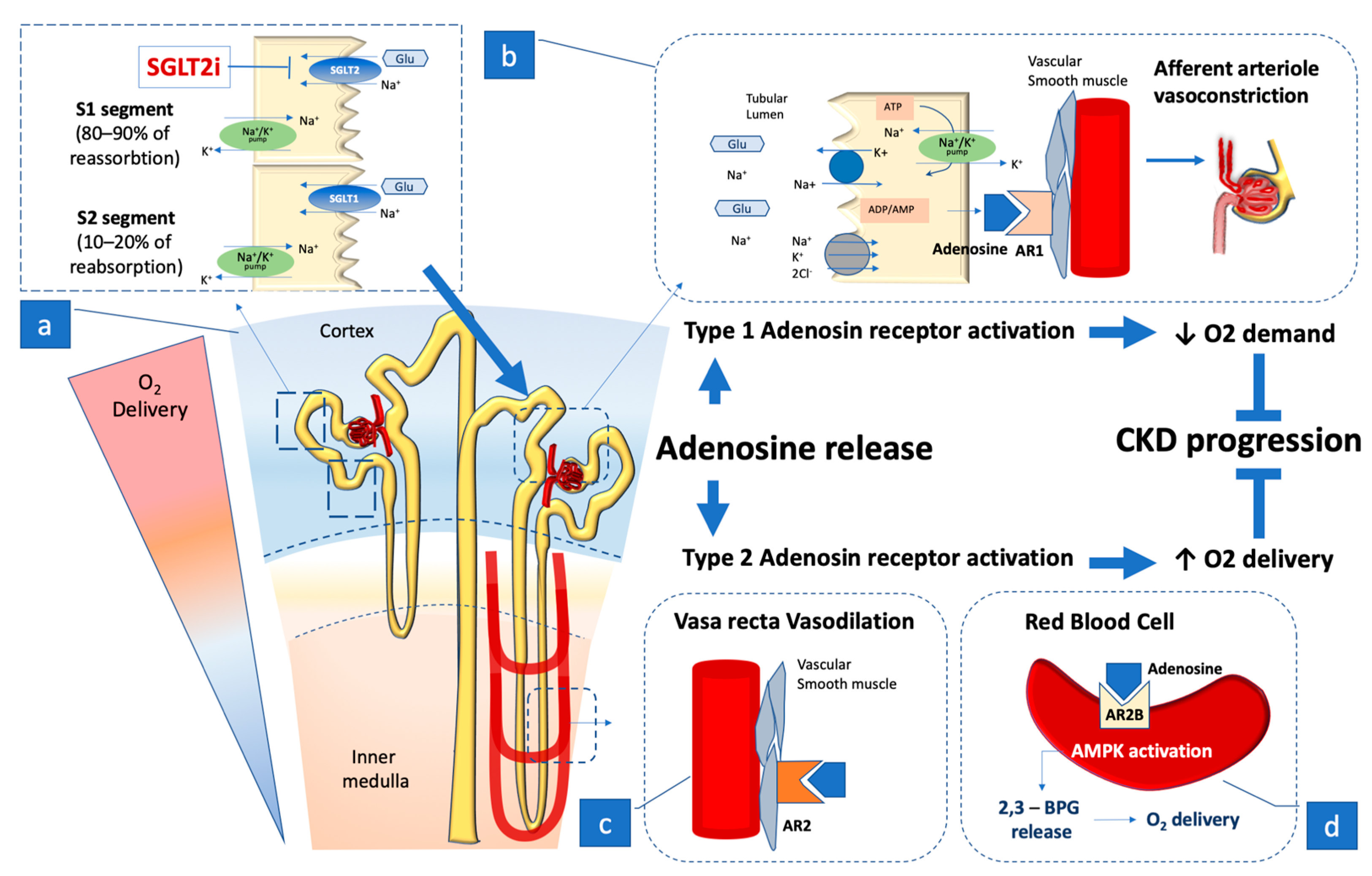
4. The Role of Adenosine
4.1. Adenosine Receptors

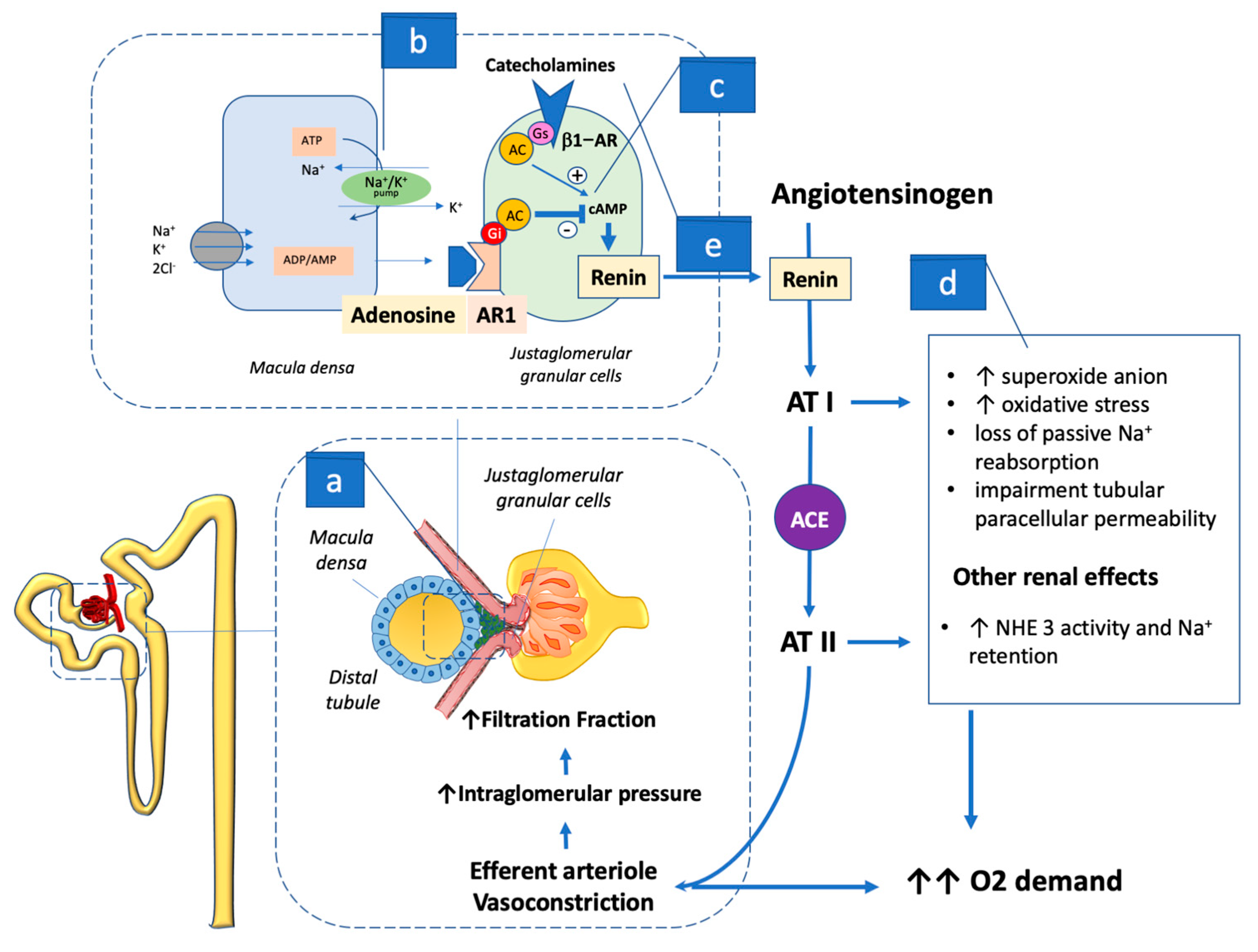
4.2. Adenosine Action, Macula Densa, and Renin–Angiotensin–Aldosterone System Activation
5. SGLT2 Activity and Sympathetic Drive
5.1. Effects on Sympathetic Activity
5.2. SGLT2 and SGLT1 Synergy and “Off Target” Implications
5.3. Other Implications of SGLT2 Inhibition and RAAS Interaction
6. SGLT2 Activity and Acute Kidney Injury
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heerspink, H.J.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: Cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 2016, 134, 752–772. [Google Scholar]
- Zinman, B.; Lachin, J.M.; Inzucchi, S.E.; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Mosenzon, O.; Wiviott, S.D.; Heerspink, H.J.; Dwyer, J.P.; Cahn, A.; Goodrich, E.L.; Rozenberg, A.; Schechter, M.; Yanuv, I.; Murphy, S.A.; et al. The Effect of Dapagliflozin on Albuminuria in DECLARE-TIMI 58. Diabetes Care 2021, 44, 1805–1815. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar]
- The EMPA-KIDNEY Collaborative Group. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2022, 388, 117–127. [Google Scholar]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canaglifozin and renal out-comes in type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardio-vascular and renal outcomes with empagliflozin in heart failure. N. Eng. J. Med. 2020, 383, 1413–1424. [Google Scholar]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. EMPEROR-Preserved Trial Investigators. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. DELIVER Trial Committees and Investigators. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Bayés-Genís, A.; Aimo, A.; Metra, M.; Anker, S.; Seferovic, P.; Rapezzi, C.; Castiglione, V.; Núñez, J.; Emdin, M.; Rosano, G.; et al. Head-to-head comparison between recommendations by the ESC and ACC/AHA/HFSA heart failure guidelines. Eur. J. Heart Fail. 2022, 24, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Schautz, B.; Later, W.; Heymsfield, S.B.; Müller, M.J. Specific metabolic rates of major organs and tissues across adulthood: Evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 2010, 92, 1369–1377. [Google Scholar] [PubMed]
- Carlström, M.; Wilcox, C.S.; Arendshorst, W.J. Renal autoregulation in health and disease. Physiol. Rev. 2015, 95, 405–511. [Google Scholar] [PubMed]
- Duncker, D.J.; Bache, R.J. Regulation of coronary blood flow during exercise. Physiol. Rev. 2008, 88, 1009–1086. [Google Scholar]
- Goodwill, A.G.; Dick, G.M.; Kiel, A.M.; Tune, J.D. Regulation of Coronary Blood Flow. Compr. Physiol. 2017, 7, 321–382. [Google Scholar]
- Saks, V.; Dzeja, P.; Schlattner, U.; Vendelin, M.; Terzic, A.; Wallimann, T. Cardiac system bioenergetics: Metabolic basis of the Frank-Starling law. J. Physiol. 2006, 571, 253–273. [Google Scholar]
- Levy, M.N. Effect of variations of blood flow on renal oxygen extraction. Am. J. Physiol. 1960, 199, 13–18. [Google Scholar] [CrossRef]
- Vallon, V.; Mühlbauer, B.; Osswald, H. Adenosine and kidney function. Physiol. Rev. 2006, 86, 901–940. [Google Scholar]
- Vallon, V. Glucose transporters in the kidney in health and disease. Pflugers. Arch. 2020, 472, 1345–1370. [Google Scholar]
- Hansell, P.; Welch, W.J.; Blantz, R.C.; Palm, F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 2013, 40, 123–137. [Google Scholar]
- Liu, Z.Z.; Bullen, A.; Li, Y.; Singh, P. Renal Oxygenation in the Pathophysiology of Chronic Kidney Disease. Front. Physiol. 2017, 8, 385. [Google Scholar] [PubMed]
- Layton, A.T.; Vallon, V. SGLT2 inhibition in a kidney with reduced nephron number: Modeling and analysis of solute transport and metabolism. Am. J. Physiol. Renal Physiol. 2018, 314, F969–F984. [Google Scholar] [PubMed]
- Jessup, M.; Costanzo, M.R. The cardiorenal syndrome: Do we need a change of strategy or a change of tactics? J. Am. Coll Cardiol. 2009, 53, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Magee, G.M.; Bilous, R.W.; Cardwell, C.R.; Hunter, S.J.; Kee, F.; Fogarty, D.G. Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta-analysis. Diabetologia 2009, 52, 691–697. [Google Scholar]
- Ruggenenti, P.; Porrini, E.L.; Gaspari, F.; Motterlini, N.; Cannata, A.; Carrara, F.; Cella, C.; Ferrari, S.; Stucchi, N.; Parvanova, A.; et al. Glomerular hyperfiltration and renal disease progression in type 2 diabetes. Diabetes Care 2012, 35, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Miracle, C.; Thomson, S. Adenosine and kidney function: Potential implications in patients with heart failure. Eur. J. Heart Fail 2008, 10, 176–187. [Google Scholar] [PubMed]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef]
- Peng, Z.; Luo, R.; Xie, T.; Zhang, W.; Liu, H.; Wang, W.; Tao, L.; Kellems, R.E.; Xia, Y. Erythrocyte Adenosine A2B Receptor-Mediated AMPK Activation: A Missing Component Counteracting CKD by Promoting Oxygen Delivery. J. Am. Soc. Nephrol. 2019, 30, 1413–1424. [Google Scholar] [CrossRef]
- Peti-Peterdi, J.; Harris, R.C. Macula densa sensing and signaling mechanisms of renin release. J. Am. Soc. Nephrol. 2010, 21, 1093–1096. [Google Scholar] [CrossRef]
- Dietrich, M.S.; Steinhausen, M. Differential reactivity of cortical and juxtaglomerullary glomeruli to adenosine-1 and adenosine-2 receptor stimulation and angiotensin converting-enzyme inhibition. Microvasc. Res. 1993, 45, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Spielman, W.S.; Osswald, H. Blockade of postocclusive renal vasoconstriction by an angiotensin II antagonists: Evidence for an angiotensin-adenosine interaction. Am. J. Physiol. 1979, 237, F463–F467. [Google Scholar] [CrossRef] [PubMed]
- Holz, F.G.; Steinhausen, M. Renovascular effects of adenosine receptor agonists. Ren. Physiol. 1987, 10, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Vander, A.J.; Wilde, W.S.; Malvin, R.L.; Sullivan, L.P. Re-examination of salt and water retention in congestive heart failure: Significance of renal filtration fraction. Am. J. Med. 1958, 25, 497–502. [Google Scholar] [CrossRef]
- Fliser, D.; Zeier, M.; Nowack, R.; Ritz, E. Renal functional reserve in healthy elderly subjects. J. Am. Soc. Nephrol. 1993, 3, 1371–1377. [Google Scholar] [CrossRef]
- Verbrugge, F.H.; Dupont, M.; Steels, P. The kidney in congestive heart failure: ‘Are natriuresis, sodium, and diuretics really the good, the bad and the ugly?’. Eur. J. Heart Fail 2014, 16, 133–142. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar]
- Gill, P.S.; Wilcox, C.S. NADPH oxidases in the kidney. Antioxid. Redox Signal. 2006, 8, 1597–1607. [Google Scholar] [CrossRef]
- Tanaka, H.; Takano, K.; Iijima, H.; Kubo, H.; Maruyama, N.; Hashimoto, T.; Arakawa, K.; Togo, M.; Inagaki, N.; Kaku, K. Factors Affecting Canagliflozin-Induced Transient Urine Volume Increase in Patients with Type 2 Diabetes Mellitus. Adv. Ther. 2017, 34, 436–451. [Google Scholar] [CrossRef]
- Sawamura, T.; Karashima, S.; Nagase, S.; Nambo, H.; Shimizu, E.; Higashitani, T.; Aono, D.; Ohbatake, A.; Kometani, M.; Demura, M.; et al. Effect of sodium-glucose cotransporter-2 inhibitors on aldosterone-to-renin ratio in diabetic patients with hypertension: A retrospective observational study. BMC Endocr. Disord. 2020, 20, 177. [Google Scholar]
- Ansary, T.M.; Nakano, D.; Nishiyama, A. Diuretic Effects of Sodium Glucose Cotransporter 2 Inhibitors and Their Influence on the Renin-Angiotensin System. Int. J. Mol. Sci. 2019, 20, 629. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Hypoxia-inducible factors in the kidney. Am. J. Physiol. Renal Physiol. 2006, 291, F271–F281. [Google Scholar] [CrossRef]
- Nespoux, J.; Patel, R.; Zhang, H.; Huang, W.; Freeman, B.; Sanders, P.W.; Kim, Y.C.; Vallon, V. Gene knockout of the Na+-glucose cotransporter SGLT2 in a murine model of acute kidney injury induced by ischemia-reperfusion. Am. J. Physiol. Renal Physiol. 2020, 318, F1100–F1112. [Google Scholar] [CrossRef] [PubMed]
- Darawshi, S.; Yaseen, H.; Gorelik, Y.; Faor, C.; Szalat, A.; Abassi, Z.; Heyman, S.N.; Khamaisi, M. Biomarker evidence for distal tubular damage but cortical sparing in hospitalized diabetic patients with acute kidney injury (AKI) while on SGLT2 inhibitors. Ren. Fail 2020, 42, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Takei, M.; Shiraishi, Y.; Suzuki, Y. Increased hematocrit during sodium-glucose cotransporter 2 inhibitor therapy indicates recovery of tubulointerstitial function in diabetic kidneys. J. Clin. Med. Res. 2016, 8, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Why do the kidneys release renin in patients with congestive heart failure? A nephrocentric view of converting-enzyme inhibition. Eur. Heart J. 1990, 11 (Suppl. D), 44–52. [Google Scholar] [CrossRef]
- Nuffield Department of Population Health Renal Studies Group; SGLT2 inhibitor Meta-Analysis Cardio-Renal Trialists’ Consortium. Impact of diabetes on the effects of sodium glucose co-transporter-2 inhibitors on kidney outcomes: Collaborative meta-analysis of large placebo-controlled trials. Lancet 2022, 400, 1788–1801. [Google Scholar] [CrossRef]
- Florea, V.G.; Cohn, J.N. The autonomic nervous system and heart failure. Circ. Res. 2014, 114, 1815–1826. [Google Scholar] [CrossRef]
- Quan, A.; Baum, M. Regulation of proximal tubule transport by endogenously produced angiotensin II. Nephron 2000, 84, 103–110. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, X.; Katsurada, K.; Patel, K.P. Renal denervation improves sodium excretion in rats with chronic heart failure: Effects on expression of renal ENaC and AQP2. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H958–H968. [Google Scholar] [CrossRef]
- Bozkurt, B.; Nair, A.P.; Misra, A.; Scott, C.Z.; Mahar, J.H.; Fedson, S. Neprilysin Inhibitors in Heart Failure: The Science, Mechanism of Action, Clinical Studies, and Unanswered Questions. JACC Basic Transl. Sci. 2022, 8, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Katsurada, K.; Nandi, S.S.; Sharma, N.M.; Patel, K.P. Enhanced expression and function of renal SGLT2 (sodium-glucose cotransporter 2) in heart failure: Role of renal nerves. Circ. Heart Fail 2021, 14, e008365. [Google Scholar] [CrossRef] [PubMed]
- Scholtes, R.A.; Muskiet, M.H.A.; van Baar, M.J.B.; Hesp, A.C.; Greasley, P.J.; Karlsson, C.; Hammarstedt, A.; Arya, N.; van Raalte, D.H.; Heerspink, H.J.L. Natriuretic Effect of Two Weeks of Dapagliflozin Treatment in Patients With Type 2 Diabetes and Preserved Kidney Function During Standardized Sodium Intake: Results of the DAPASALT Trial. Diabetes Care 2021, 44, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Herat, L.Y.; Magno, A.L.; Rudnicka, C.; Hricova, J.; Carnagarin, R.; Ward, N.C.; Arcambal, A.; Kiuchi, M.G.; Head, G.A.; Schlaich, M.P.; et al. SGLT2 inhibitor-induced sympatho-inhibition: A novel mechanism for cardiorenal protection. JACC Basic Transl. Sci. 2020, 5, 169–179. [Google Scholar] [CrossRef]
- Hasking, G.J.; Esler, M.D.; Jennings, G.L.; Burton, D.; Johns, J.A.; Korner, P.I. Norepinephrine spillover to plasma in patients with congestive heart failure: Evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation 1986, 73, 615–621. [Google Scholar] [CrossRef]
- Petersson, M.; Friberg, P.; Eisenhofer, G.; Lambert, G.; Rundqvist, B. Long-term outcome in relation to renal sympathetic activity in patients with chronic heart failure. Eur. Heart J. 2005, 26, 906–913. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; DeFronzo, R.A.; Norton, L. Novel hypothesis to explain why SGLT2 inhibitors inhibit only 30-50% of filtered glucose load in humans. Diabetes 2013, 62, 3324–3328. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, J.; Jiang, S.; Xu, L.; Wang, L.; Cheng, F.; Buggs, J.; Koepsell, H.; Vallon, V.; Liu, R. Macula Densa SGLT1-NOS1-Tubuloglomerular Feedback Pathway, a New Mechanism for Glomerular Hyperfiltration during Hyperglycemia. J. Am. Soc. Nephrol. 2019, 30, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Huang, W.; Onishi, A.; Patel, R.; Kim, Y.C.; van Ginkel, C.; Fu, Y.; Freeman, B.; Koepsell, H.; Thomson, S.; et al. Knockout of Na+-glucose cotransporter SGLT1 mitigates diabetes-induced upregulation of nitric oxide synthase NOS1 in the macula densa and glomerular hyperfiltration. Am. J. Physiol. Renal Physiol. 2019, 317, F207–F217. [Google Scholar] [CrossRef]
- Packer, M. Role of Impaired Nutrient and Oxygen Deprivation Signaling and Deficient Autophagic Flux in Diabetic CKD Development: Implications for Understanding the Effects of Sodium-Glucose Cotransporter 2-Inhibitors. J. Am. Soc. Nephrol. 2020, 31, 907–919. [Google Scholar] [CrossRef]
- Onishi, A.; Fu, Y.; Patel, R.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Freeman, B.; Kim, Y.C.; Soleimani, M.; et al. A role for tubular Na+/H+ exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol. Renal Physiol. 2020, 319, F712–F728. [Google Scholar] [CrossRef]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes. Metab. 2018, 20, 479–487. [Google Scholar] [CrossRef]
- Griffin, M.; Rao, V.S.; Ivey-Miranda, J.; Fleming, J.; Mahoney, D.; Maulion, C.; Suda, N.; Siwakoti, K.; Ahmad, T.; Jacoby, D.; et al. Empagliflozin in Heart Failure: Diuretic and Cardiorenal Effects. Circulation 2020, 142, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S.; Shen, W.; Boulton, D.W.; Leslie, B.R.; Griffen, S.C. Interaction Between the Sodium-Glucose-Linked Transporter 2 Inhibitor Dapagliflozin and the Loop Diuretic Bumetanide in Normal Human Subjects. J. Am. Heart Assoc. 2018, 7, e007046. [Google Scholar] [CrossRef] [PubMed]
- Eickhoff, M.K.; Dekkers, C.C.J.; Kramers, B.J.; Laverman, G.D.; Frimodt-Møller, M.; Jørgensen, N.R.; Faber, J.; Danser, A.H.J.; Gansevoort, R.T.; Rossing, P.; et al. Effects of dapagliflozin on volume status when added to renin angiotensin system inhibitors. J. Clin. Med. 2019, 8, 779. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Watanabe, Y.; Fukuda, K.; Watanabe, M.; Onishi, A.; Ohara, K.; Imai, T.; Koepsell, H.; Muto, S.; Vallon, V.; et al. Unmasking a sustained negative effect of SGLT2 inhibition on body fluid volume in the rat. Am. J. Physiol. Ren. Physiol. 2018, 315, F653–F664. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Muto, S.; Fukuda, K.; Watanabe, M.; Ohara, K.; Koepsell, H.; Vallon, V.; Nagata, D. Osmotic diuresis by SGLT2 inhibition stimulates vasopressin-induced water reabsorption to maintain body fluid volume. Physiol. Rep. 2020, 8, e14360. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Bjornstad, P.; Katz, A.; Singh, S.; Godoy, L.; Chung, L.; Vinovskis, C.; Pyle, L.; Roussel, R.; Perkins, B.; et al. SGLT2 inhibition increases serum copeptin in young adults with type 1 diabetes. Diabetes Metab. 2020, 46, 203–209. [Google Scholar] [CrossRef]
- Suijk, D.L.S.; van Baar, M.J.B.; van Bommel, E.J.M.; Iqbal, Z.; Krebber, M.M.; Vallon, V.; Touw, D.; Hoorn, E.J.; Nieuwdorp, M.; Kramer, M.M.H.; et al. SGLT2 Inhibition and Uric Acid Excretion in Patients with Type 2 Diabetes and Normal Kidney Function. Clin. J. Am. Soc. Nephrol. 2022, 17, 663–671. [Google Scholar] [CrossRef]
- Hahn, K.; Ejaz, A.A.; Kanbay, M.; Lanaspa, M.A.; Johnson, R.J. Acute kidney injury from SGLT2 inhibitors: Potential mechanisms. Nat. Rev. Nephrol. 2016, 12, 711–712. [Google Scholar] [CrossRef]
- Hahn, K.; Kanbay, M.; Lanaspa, M.A.; Johnson RJ Ejaz, A.A. Serum uric acid and acute kidney injury: A mini review. J. Adv. Res. 2017, 8, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Bjornstad, P.; Lanaspa, M.A.; Ishimoto, T. Fructose and uric acid in diabetic nephropathy. Diabetologia 2015, 58, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Yamauchi, A.; Sugiura, T. Inhibition of myo-inositol transport causes acute renal failure with selective medullary injury in the rat. Kidney Int. 1998, 53, 146–153. [Google Scholar] [CrossRef]
- Laursen, J.C.; Søndergaard-Heinrich, N.; de Melo, J.M.L.; Haddock, B.; Rasmussen, I.K.B.; Safavimanesh, F.; Hansen, C.S.; Størling, J.; Larsson, H.B.W.; Groop, P.-H.; et al. Acute effects of dapagliflozin on renal oxygenation and perfusion in type 1 diabetes with albuminuria: A randomised, double-blind, placebo-controlled crossover trial. EClinicalMedicine 2021, 37, 100895. [Google Scholar] [CrossRef] [PubMed]
- Neuen, B.L.; Young, T.; Heerspink, H.J.L.; Neal, B.; Perkovic, V.; Billot, L.; Mahaffey, K.W.; Charytan, D.M.; Wheeler, D.C.; Arnott, C.; et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019, 7, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Cahn, A.; Melzer-Cohen, C.; Pollack, R.; Chodick, G.; Shalev, V. Acute renal outcomes with sodium-glucose co-transporter-2 inhibitors: Real-world data analysis. Diabetes Obes. Metab. 2019, 21, 340–348. [Google Scholar] [CrossRef]
- Iskander, C.; Cherney, D.Z.; Clemens, K.K.; Dixon, S.N.; Harel, Z.; Jeyakumar, N.; McArthur, E.; Muanda, F.T.; Parikh, C.R.; Paterson, J.M.; et al. Use of sodium-glucose cotransporter-2 inhibitors and risk of acute kidney injury in older adults with diabetes: A population-based cohort study. CMAJ 2020, 192, E351–E360. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gronda, E.; Palazzuoli, A.; Iacoviello, M.; Benevenuto, M.; Gabrielli, D.; Arduini, A. Renal Oxygen Demand and Nephron Function: Is Glucose a Friend or Foe? Int. J. Mol. Sci. 2023, 24, 9957. https://doi.org/10.3390/ijms24129957
Gronda E, Palazzuoli A, Iacoviello M, Benevenuto M, Gabrielli D, Arduini A. Renal Oxygen Demand and Nephron Function: Is Glucose a Friend or Foe? International Journal of Molecular Sciences. 2023; 24(12):9957. https://doi.org/10.3390/ijms24129957
Chicago/Turabian StyleGronda, Edoardo, Alberto Palazzuoli, Massimo Iacoviello, Manuela Benevenuto, Domenico Gabrielli, and Arduino Arduini. 2023. "Renal Oxygen Demand and Nephron Function: Is Glucose a Friend or Foe?" International Journal of Molecular Sciences 24, no. 12: 9957. https://doi.org/10.3390/ijms24129957
APA StyleGronda, E., Palazzuoli, A., Iacoviello, M., Benevenuto, M., Gabrielli, D., & Arduini, A. (2023). Renal Oxygen Demand and Nephron Function: Is Glucose a Friend or Foe? International Journal of Molecular Sciences, 24(12), 9957. https://doi.org/10.3390/ijms24129957