
Multiplatform Metabolomics Characterization Reveals Novel Metabolites and Phospholipid Compositional Rules of Haemophilus influenzae Rd KW20

 , , , ,
, , , ,
Abstract
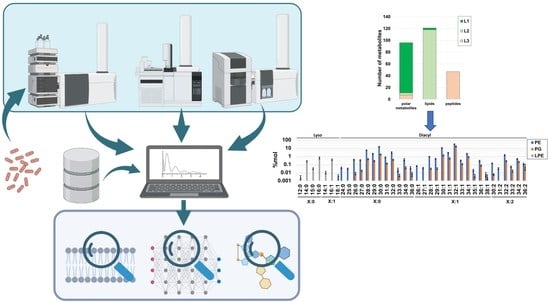
:
1. Introduction
2. Results and Discussion
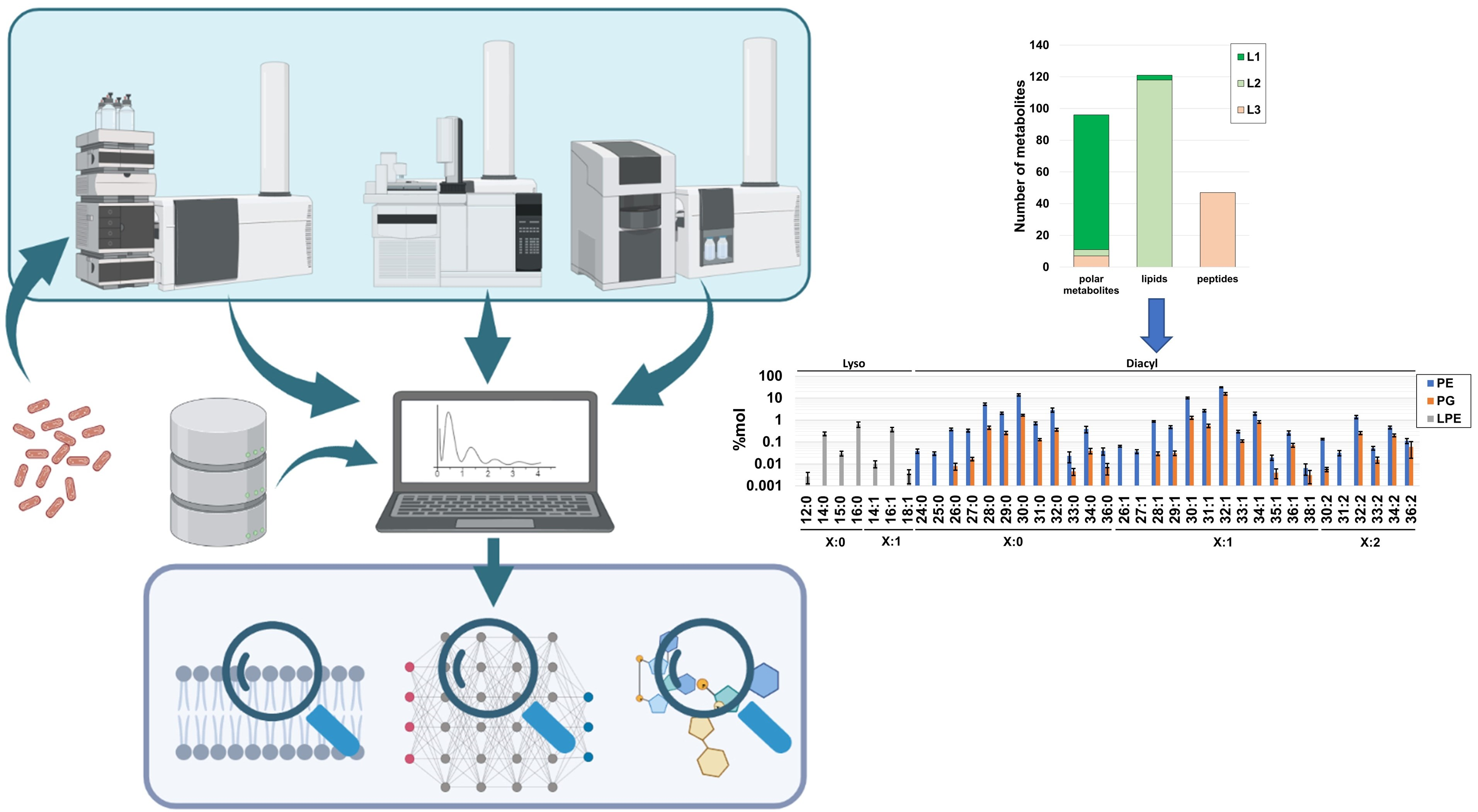
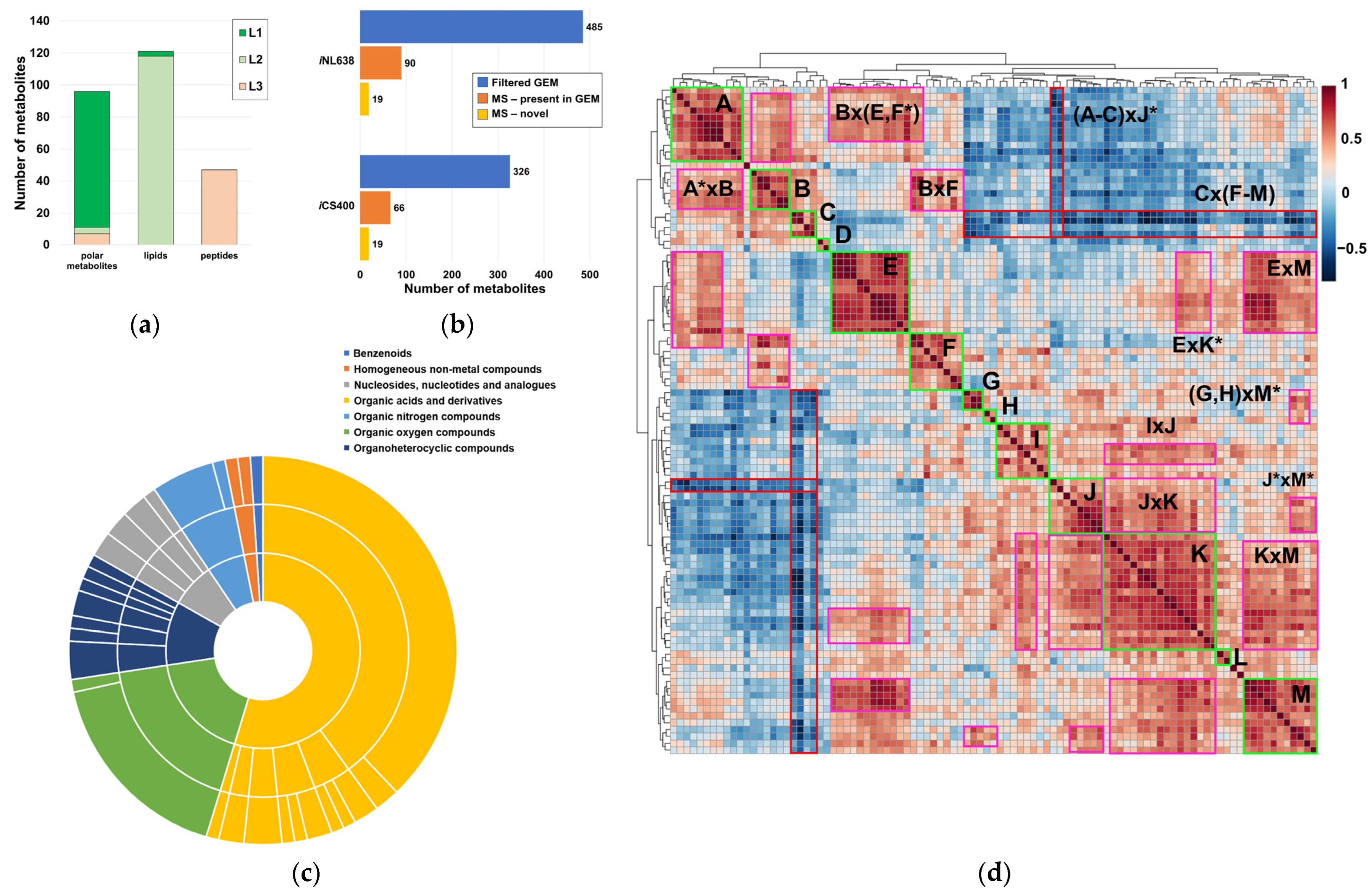
2.1. Global Compositional Properties of the Experimentally Determined Metabolome of Haemophilus influenzae
2.2. Experimental Polar Metabolome Characterization Reveals Novel Metabolites of H. influenzae
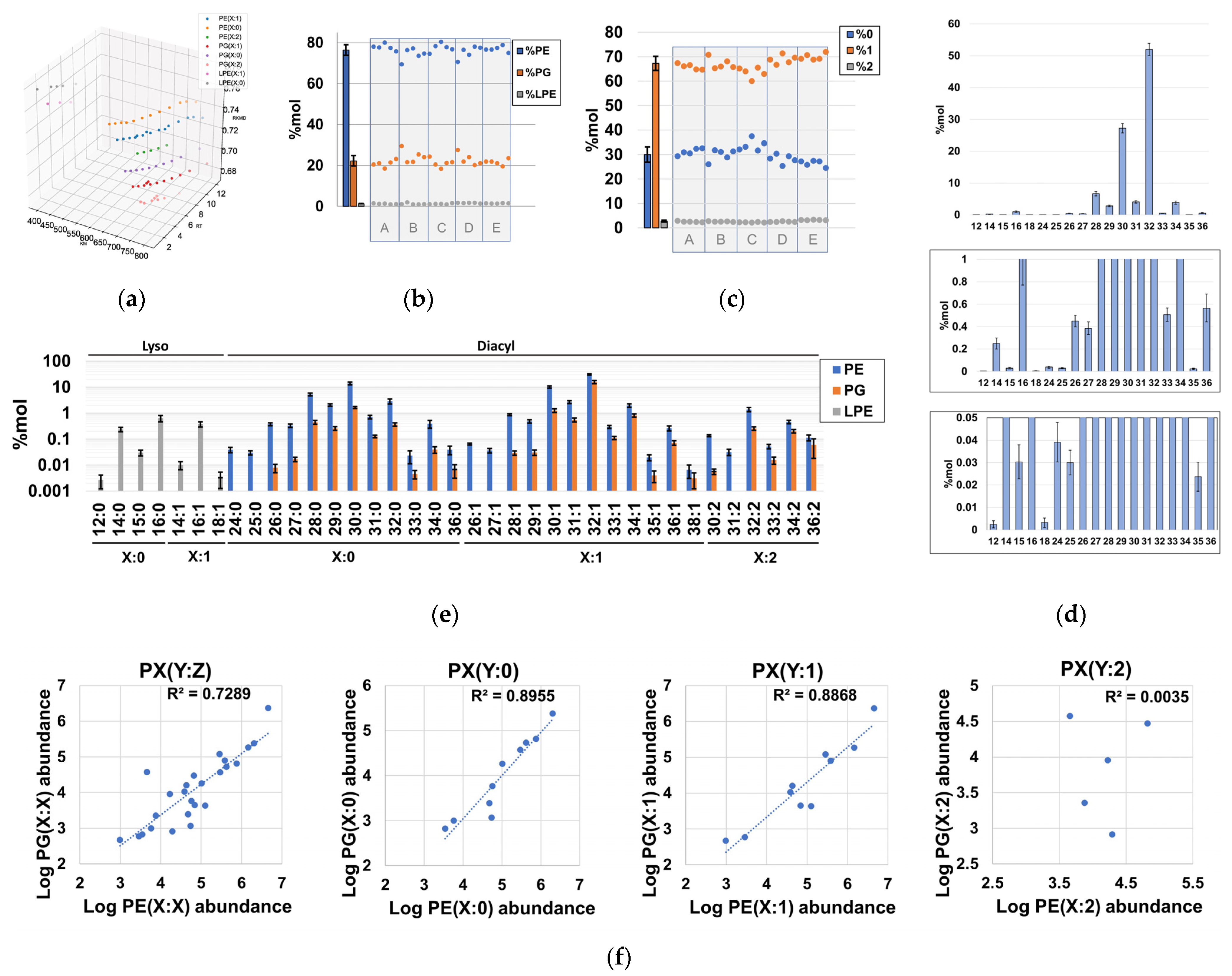
2.3. Global Compositional Properties of the Phospholipidome of H. influenzae
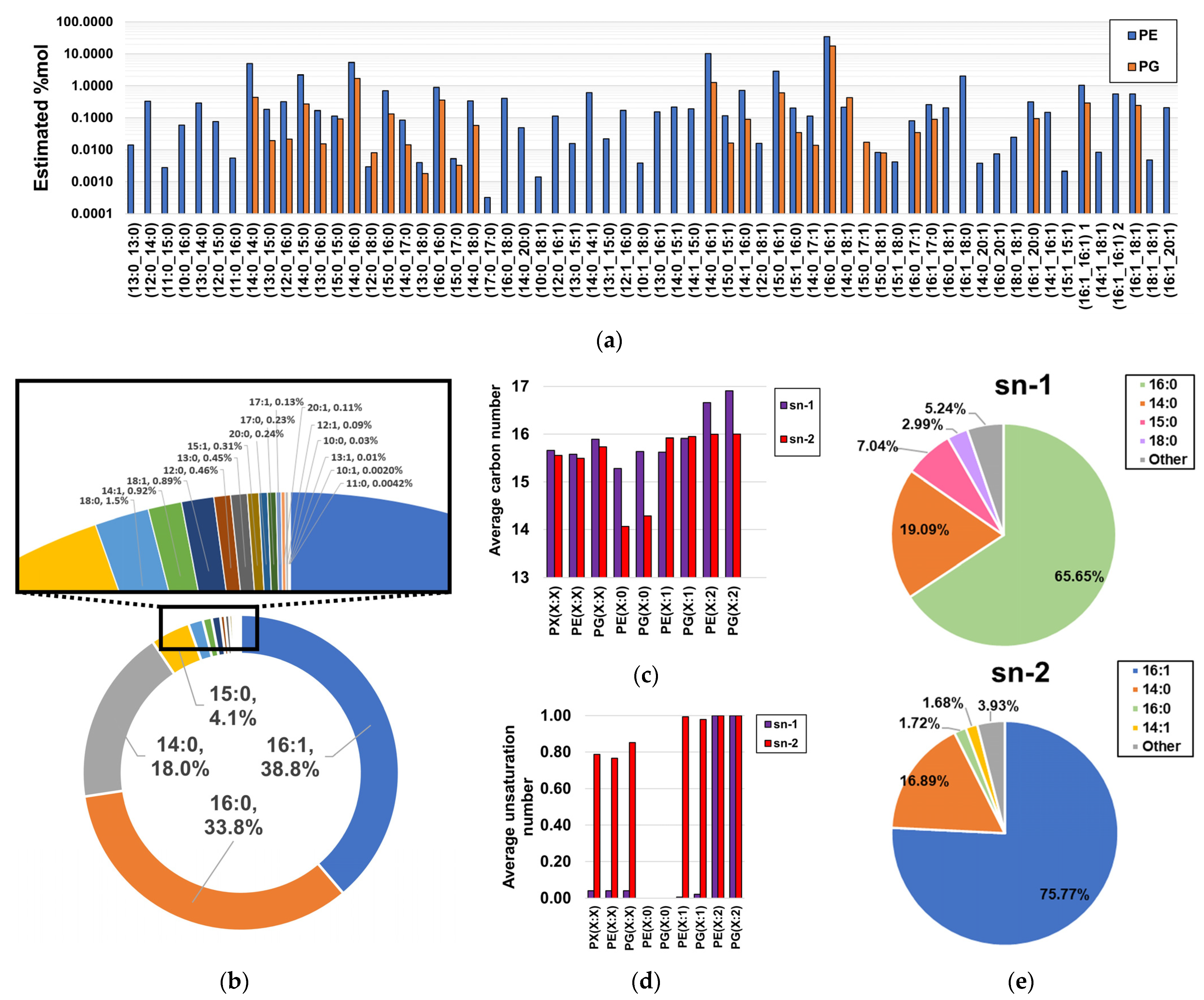
2.4. Fragmentation Analysis of Phospholipid Coelutions Allow for a Detailed Estimation of the Fatty Acyl Composition in the H. influenzae Lipidome and Positional Information of Fatty Acyl Chains in Diacylglycerophospholipids
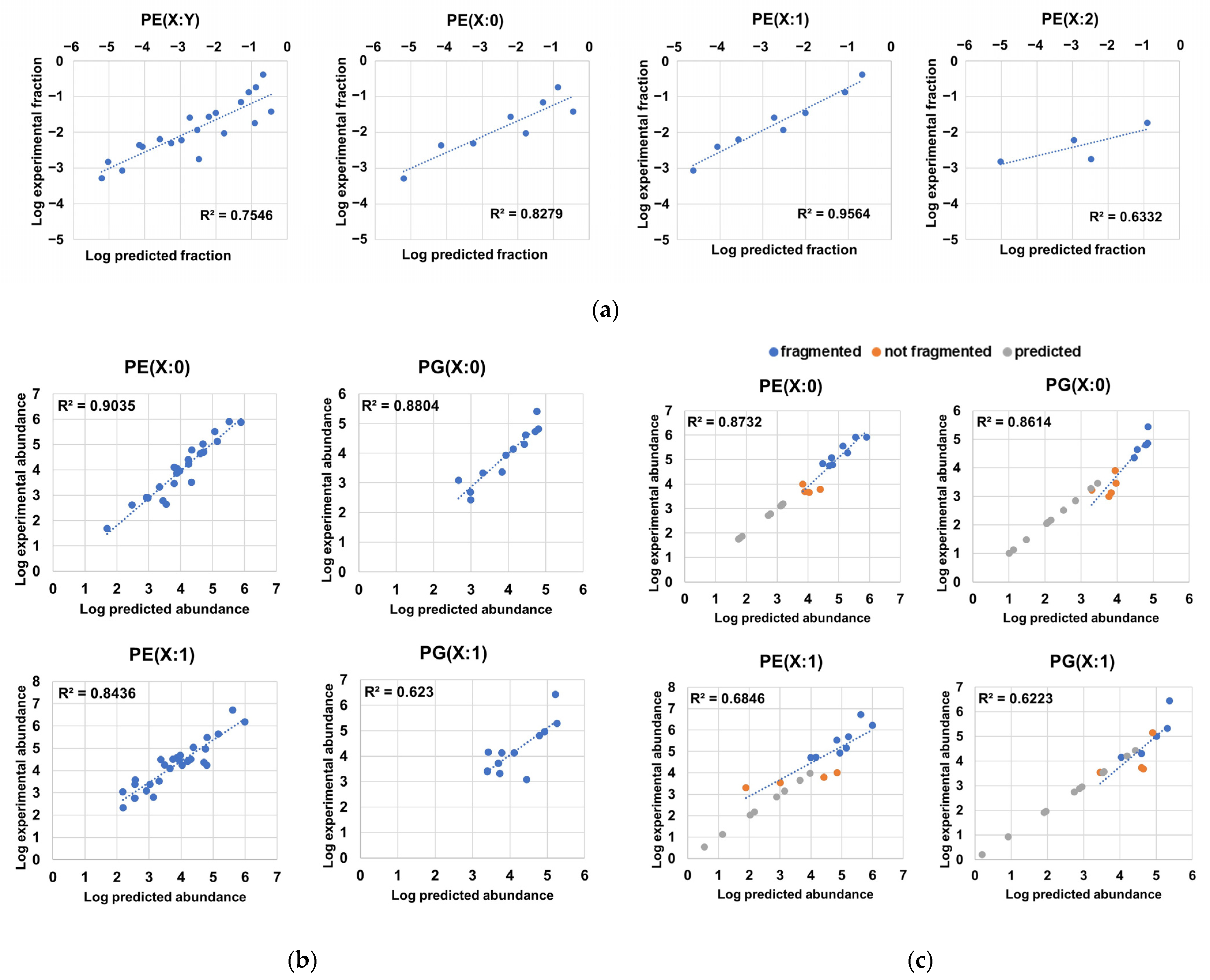
2.5. Probabilistic Rules Are a Major Factor Defining the Order of Magnitude of Lipid Species, Allowing the Prediction of Low-Level Species, Expansion and Refinement of the Phospholipidome
3. Materials and Methods
3.1. Reagents and Solutions
3.2. Bacterial Strains and Culture Conditions
3.3. Cellular Lysis and Metabolite Extraction
3.4. LC-QTOF/MS Analysis and Data Processing
3.5. GC-QTOF/MS Analysis and Data Processing
3.6. CE-TOF/MS Analysis and Data Processing
3.7. Curation and Obtention of the Small Molecule Set of iCS400 and iNL638 Metabolic Models
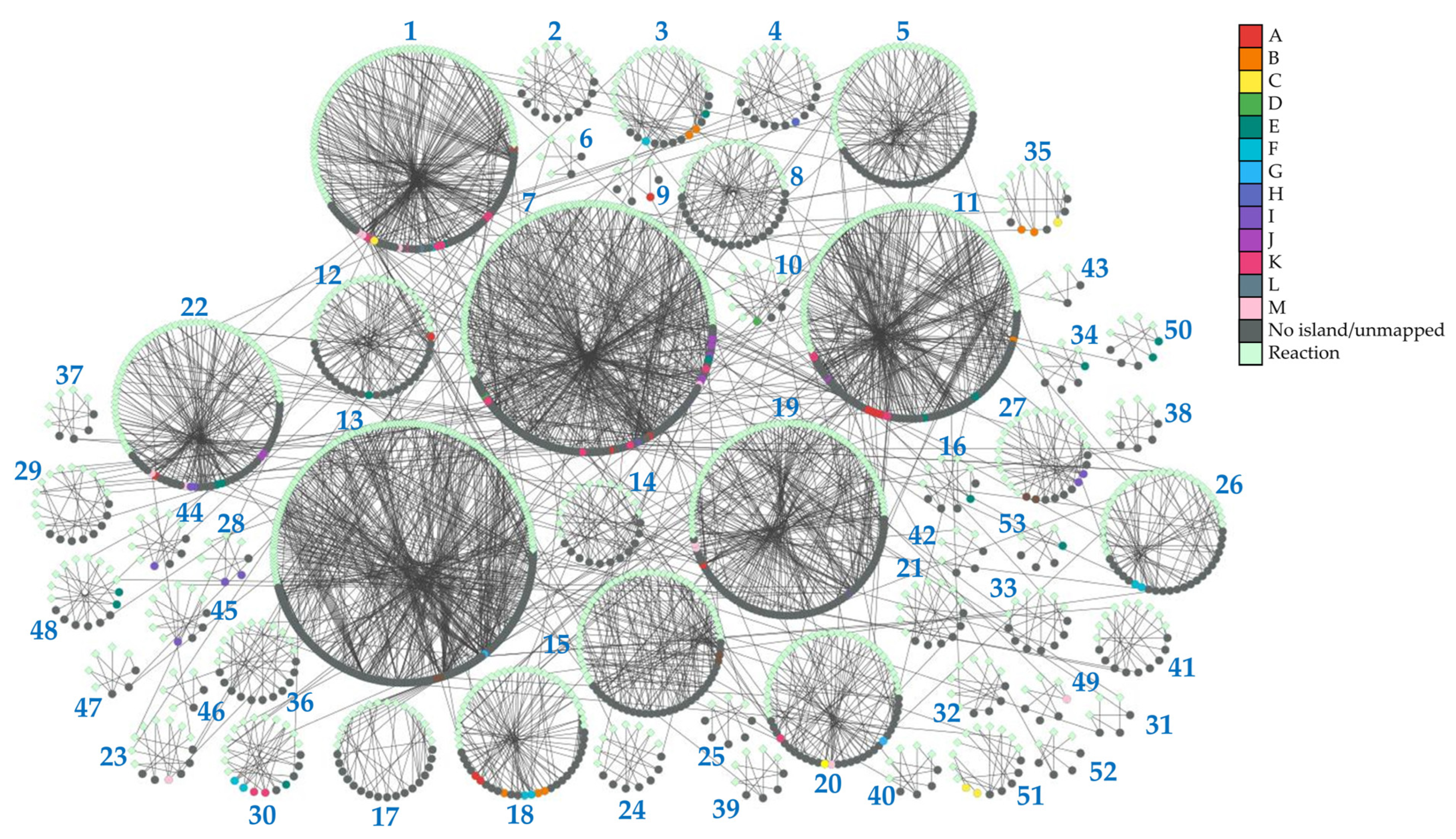
3.8. Curation and Network Analysis of the Small Molecule Set of iCS400 and iNL638 Metabolic Models
3.9. Global Evaluation of Metabolite Experimental Data
3.10. Calculation of Experimental Lipidomic Properties
3.11. Calculation of Predictive Lipidomic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agrawal, A.; Murphy, T.F. Haemophilus influenzae infections in the H. influenzae type b conjugate vaccine era. J. Clin. Microbiol. 2011, 49, 3728–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaClaire, L.L.; Tondella, M.L.; Beall, D.S.; Noble, C.A.; Raghunathan, P.L.; Rosenstein, N.E.; Popovic, T.; Members, A.B.C.S.T. Identification of Haemophilus influenzae serotypes by standard slide agglutination serotyping and PCR-based capsule typing. J. Clin. Microbiol. 2003, 41, 393–396. [Google Scholar] [CrossRef] [Green Version]
- Khattak, Z.E.; Anjum, F. Haemophilus Influenzae. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Wahl, B.; O’Brien, K.L.; Greenbaum, A.; Majumder, A.; Liu, L.; Chu, Y.; Lukšić, I.; Nair, H.; McAllister, D.A.; Campbell, H.; et al. Burden of Streptococcus pneumoniae and Haemophilus influenzae type b disease in children in the era of conjugate vaccines: Global, regional, and national estimates for 2000–15. Lancet Glob. Health 2018, 6, e744–e757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langereis, J.D.; de Jonge, M.I. Invasive Disease Caused by Nontypeable Haemophilus influenzae. Emerg. Infect. Dis. 2015, 21, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Van Eldere, J.; Slack, M.P.; Ladhani, S.; Cripps, A.W. Non-typeable Haemophilus influenzae, an under-recognised pathogen. Lancet Infect. Dis. 2014, 14, 1281–1292. [Google Scholar] [CrossRef] [Green Version]
- Behrouzi, A.; Vaziri, F.; Rahimi-Jamnani, F.; Afrough, P.; Rahbar, M.; Satarian, F.; Siadat, S.D. Vaccine Candidates against Nontypeable Haemophilus influenzae: A Review. Iran. Biomed. J. 2017, 21, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Poje, G.; Redfield, R.J. General methods for culturing Haemophilus influenzae. Methods Mol. Med. 2003, 71, 51–56. [Google Scholar] [CrossRef]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [Green Version]
- Baddal, B.; Muzzi, A.; Censini, S.; Calogero, R.A.; Torricelli, G.; Guidotti, S.; Taddei, A.R.; Covacci, A.; Pizza, M.; Rappuoli, R.; et al. Dual RNA-seq of Nontypeable Haemophilus influenzae and Host Cell Transcriptomes Reveals Novel Insights into Host-Pathogen Cross Talk. mBio 2015, 6, e01765-15. [Google Scholar] [CrossRef] [Green Version]
- Link, A.J.; Hays, L.G.; Carmack, E.B.; Yates, J.R. Identifying the major proteome components of Haemophilus influenzae type-strain NCTC 8143. Electrophoresis 1997, 18, 1314–1334. [Google Scholar] [CrossRef]
- Qu, J.; Lesse, A.J.; Brauer, A.L.; Cao, J.; Gill, S.R.; Murphy, T.F. Proteomic expression profiling of Haemophilus influenzae grown in pooled human sputum from adults with chronic obstructive pulmonary disease reveal antioxidant and stress responses. BMC Microbiol. 2010, 10, 162. [Google Scholar] [CrossRef] [Green Version]
- Kolker, E.; Purvine, S.; Galperin, M.Y.; Stolyar, S.; Goodlett, D.R.; Nesvizhskii, A.I.; Keller, A.; Xie, T.; Eng, J.K.; Yi, E.; et al. Initial proteome analysis of model microorganism Haemophilus influenzae strain Rd KW20. J. Bacteriol. 2003, 185, 4593–4602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Post, D.M.; Held, J.M.; Ketterer, M.R.; Phillips, N.J.; Sahu, A.; Apicella, M.A.; Gibson, B.W. Comparative analyses of proteins from Haemophilus influenzae biofilm and planktonic populations using metabolic labeling and mass spectrometry. BMC Microbiol. 2014, 14, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.S.; Palsson, B.O. Systems properties of the Haemophilus influenzae Rd metabolic genotype. J. Biol. Chem. 1999, 274, 17410–17416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Othman, D.S.; Schirra, H.; McEwan, A.G.; Kappler, U. Metabolic versatility in Haemophilus influenzae: A metabolomic and genomic analysis. Front. Microbiol. 2014, 5, 69. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Mushegian, A.R.; Bork, P.; Brown, N.P.; Hayes, W.S.; Borodovsky, M.; Rudd, K.E.; Koonin, E.V. Metabolism and evolution of Haemophilus influenzae deduced from a whole-genome comparison with Escherichia coli. Curr. Biol. 1996, 6, 279–291. [Google Scholar] [CrossRef] [Green Version]
- López-López, N.; León, D.S.; de Castro, S.; Díez-Martínez, R.; Iglesias-Bexiga, M.; Camarasa, M.J.; Menéndez, M.; Nogales, J.; Garmendia, J. Interrogation of Essentiality in the Reconstructed Haemophilus influenzae Metabolic Network Identifies Lipid Metabolism Antimicrobial Targets: Preclinical Evaluation of a FabH β-Ketoacyl-ACP Synthase Inhibitor. mSystems 2022, 7, e0145921. [Google Scholar] [CrossRef]
- Schilling, C.H.; Palsson, B.O. Assessment of the metabolic capabilities of Haemophilus influenzae Rd through a genome-scale pathway analysis. J. Theor. Biol. 2000, 203, 249–283. [Google Scholar] [CrossRef]
- López-López, N.; Euba, B.; Hill, J.; Dhouib, R.; Caballero, L.A.; Leiva, J.; Hosmer, J.; Cuesta, S.; Ramos-Vivas, J.; Díez-Martínez, R.; et al. Glucose Catabolism Leading to Production of the Immunometabolite Acetate Has a Key Contribution to the Host Airway-Pathogen Interplay. ACS Infect. Dis. 2020, 6, 406–421. [Google Scholar] [CrossRef]
- Muda, N.M.; Nasreen, M.; Dhouib, R.; Hosmer, J.; Hill, J.; Mahawar, M.; Schirra, H.J.; McEwan, A.G.; Kappler, U. Metabolic analyses reveal common adaptations in two invasive Haemophilus influenzae strains. Pathog. Dis. 2019, 77, ftz015. [Google Scholar] [CrossRef]
- Ares-Arroyo, M.; Fernández-García, M.; Wedel, E.; Montero, N.; Barbas, C.; Rey-Stolle, M.F.; Garcia, A.; González-Zorn, B. Genomics, Transcriptomics, and Metabolomics Reveal That Minimal Modifications in the Host Are Crucial for the Compensatory Evolution of ColE1-Like Plasmids. mSphere 2022, 7, e0018422. [Google Scholar] [CrossRef] [PubMed]
- May, P.; Wienkoop, S.; Kempa, S.; Usadel, B.; Christian, N.; Rupprecht, J.; Weiss, J.; Recuenco-Munoz, L.; Ebenhöh, O.; Weckwerth, W.; et al. Metabolomics-and proteomics-assisted genome annotation and analysis of the draft metabolic network of Chlamydomonas reinhardtii. Genetics 2008, 179, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchlis, V.D.; Armando, A.; Dennis, E.A. Substrate-Specific Inhibition Constants for Phospholipase A. J. Med. Chem. 2019, 62, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.D.; Köhler, N.; Falk, L.; Klischat, L.; Lazareva, O.E.; Pauling, J.K. Lipid network and moiety analysis for revealing enzymatic dysregulation and mechanistic alterations from lipidomics data. Brief. Bioinform. 2023, 24, bbac572. [Google Scholar] [CrossRef]
- Tang, X.; Chang, S.; Qiao, W.; Luo, Q.; Chen, Y.; Jia, Z.; Coleman, J.; Zhang, K.; Wang, T.; Zhang, Z.; et al. Structural insights into outer membrane asymmetry maintenance in Gram-negative bacteria by MlaFEDB. Nat. Struct. Mol. Biol. 2021, 28, 81–91. [Google Scholar] [CrossRef]
- Bogdanov, M.; Pyrshev, K.; Yesylevskyy, S.; Ryabichko, S.; Boiko, V.; Ivanchenko, P.; Kiyamova, R.; Guan, Z.; Ramseyer, C.; Dowhan, W. Phospholipid distribution in the cytoplasmic membrane of Gram-negative bacteria is highly asymmetric, dynamic, and cell shape-dependent. Sci. Adv. 2020, 6, eaaz6333. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [Green Version]
- Schweda, E.K.; Richards, J.C.; Hood, D.W.; Moxon, E.R. Expression and structural diversity of the lipopolysaccharide of Haemophilus influenzae: Implication in virulence. Int. J. Med. Microbiol. 2007, 297, 297–306. [Google Scholar] [CrossRef]
- Mikhail, I.; Yildirim, H.H.; Lindahl, E.C.; Schweda, E.K. Structural characterization of lipid A from nontypeable and type f Haemophilus influenzae: Variability of fatty acid substitution. Anal. Biochem. 2005, 340, 303–316. [Google Scholar] [CrossRef]
- Sohlenkamp, C.; Geiger, O. Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev. 2016, 40, 133–159. [Google Scholar] [CrossRef] [Green Version]
- Sutrina, S.L.; Scocca, J.J. Phospholipids of Haemophilus influenzae Rd during exponential growth and following the development of competence for genetic transformation. J. Gen. Microbiol. 1976, 92, 410–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murzyn, K.; Róg, T.; Pasenkiewicz-Gierula, M. Phosphatidylethanolamine-phosphatidylglycerol bilayer as a model of the inner bacterial membrane. Biophys. J. 2005, 88, 1091–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leekumjorn, S.; Cho, H.J.; Wu, Y.; Wright, N.T.; Sum, A.K.; Chan, C. The role of fatty acid unsaturation in minimizing biophysical changes on the structure and local effects of bilayer membranes. Biochim. Biophys. Acta 2009, 1788, 1508–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2014. [Google Scholar]
- Fernández-Calvet, A.; Rodríguez-Arce, I.; Almagro, G.; Moleres, J.; Euba, B.; Caballero, L.; Martí, S.; Ramos-Vivas, J.; Bartholomew, T.L.; Morales, X.; et al. Modulation of Haemophilus influenzae interaction with hydrophobic molecules by the VacJ/MlaA lipoprotein impacts strongly on its interplay with the airways. Sci. Rep. 2018, 8, 6872. [Google Scholar] [CrossRef] [Green Version]
- Jaisinghani, N.; Seeliger, J.C. Recent advances in the mass spectrometric profiling of bacterial lipids. Curr. Opin. Chem. Biol. 2021, 65, 145–153. [Google Scholar] [CrossRef]
- Cao, W.; Cheng, S.; Yang, J.; Feng, J.; Zhang, W.; Li, Z.; Chen, Q.; Xia, Y.; Ouyang, Z.; Ma, X. Large-scale lipid analysis with C=C location and sn-position isomer resolving power. Nat. Commun. 2020, 11, 375. [Google Scholar] [CrossRef] [Green Version]
- Jeucken, A.; Molenaar, M.R.; van de Lest, C.H.A.; Jansen, J.W.A.; Helms, J.B.; Brouwers, J.F. A Comprehensive Functional Characterization of Escherichia coli Lipid Genes. Cell Rep. 2019, 27, 1597–1606.e1592. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.M.; Rock, C.O. Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 2008, 6, 222–233. [Google Scholar] [CrossRef]
- Seltmann, G.; Holst, O. The Bacterial Cell Wall, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Rakusanova, S.; Fiehn, O.; Cajka, T. Toward building mass spectrometry-based metabolomics and lipidomics atlases for biological and clinical research. TrAC Trends Anal. Chem. 2023, 158, 116825. [Google Scholar] [CrossRef]
- Frainay, C.; Schymanski, E.L.; Neumann, S.; Merlet, B.; Salek, R.M.; Jourdan, F.; Yanes, O. Mind the Gap: Mapping Mass Spectral Databases in Genome-Scale Metabolic Networks Reveals Poorly Covered Areas. Metabolites 2018, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Begum Ahil, S.; Hira, K.; Shaik, A.B.; Pal, P.P.; Kulkarni, O.P.; Araya, H.; Fujimoto, Y. l-Proline-based-cyclic dipeptides from Pseudomonas sp. (ABS-36) inhibit pro-inflammatory cytokines and alleviate crystal-induced renal injury in mice. Int. Immunopharmacol. 2019, 73, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Shao, Z.; Cai, M.; Zheng, L.; Li, G.; Yu, Z.; Zhang, J. Cyclo(l-Pro–l-Leu) of Pseudomonas putida MCCC1A00316 Isolated from Antarctic Soil: Identification and Characterization of Activity against Meloidogyne incognita. Molecules 2019, 24, 768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Huiru, Z.; Biting, D.; Yun, J.; Wei, J.; Kunming, D. Study on the anti-quorum sensing activity of a marine bacterium Staphylococcus saprophyticus 108. BTAIJ 2013, 7, 480–487. [Google Scholar]
- Parasuraman, P.; Devadatha, B.; Sarma, V.V.; Ranganathan, S.; Ampasala, D.R.; Reddy, D.; Kumavath, R.; Kim, I.W.; Patel, S.K.S.; Kalia, V.C.; et al. Inhibition of Microbial Quorum Sensing Mediated Virulence Factors by. J. Microbiol. Biotechnol. 2020, 30, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, S.; Sivaranjani, M.; Kamaladevi, A.; Ravi, A.V.; Balamurugan, K.; Karutha Pandian, S. Cyclic dipeptide cyclo(l-leucyl-l-prolyl) from marine Bacillus amyloliquefaciens mitigates biofilm formation and virulence in Listeria monocytogenes. Pathog. Dis. 2016, 74, ftw017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesan, J.T.; Morelli, T.; Moss, K.; Barros, S.P.; Ward, M.; Jenkins, W.; Aspiras, M.B.; Offenbacher, S. Association of Synergistetes and Cyclodipeptides with Periodontitis. J. Dent. Res. 2015, 94, 1425–1431. [Google Scholar] [CrossRef]
- Cerneckis, J.; Cui, Q.; He, C.; Yi, C.; Shi, Y. Decoding pseudouridine: An emerging target for therapeutic development. Trends Pharmacol. Sci. 2022, 43, 522–535. [Google Scholar] [CrossRef]
- Preumont, A.; Snoussi, K.; Stroobant, V.; Collet, J.F.; Van Schaftingen, E. Molecular identification of pseudouridine-metabolizing enzymes. J. Biol. Chem. 2008, 283, 25238–25246. [Google Scholar] [CrossRef] [Green Version]
- Thapa, K.; Oja, T.; Metsä-Ketelä, M. Molecular evolution of the bacterial pseudouridine-5’-phosphate glycosidase protein family. FEBS J. 2014, 281, 4439–4449. [Google Scholar] [CrossRef]
- Vergauwen, B.; Elegheert, J.; Dansercoer, A.; Devreese, B.; Savvides, S.N. Glutathione import in Haemophilus influenzae Rd is primed by the periplasmic heme-binding protein HbpA. Proc. Natl. Acad. Sci. USA 2010, 107, 13270–13275. [Google Scholar] [CrossRef]
- Lin, M.C.; Wagner, C. Purification and characterization of N-methylalanine dehydrogenase. J. Biol. Chem. 1975, 250, 3746–3751. [Google Scholar] [CrossRef] [PubMed]
- Hamana, K.; Nakata, K. Distribution of diaminopropane, putrescine and cadaverine in Haemophilus and Actinobacillus. Microbios 2000, 103, 43–51. [Google Scholar] [PubMed]
- Erwin, A.L.; Nelson, K.L.; Mhlanga-Mutangadura, T.; Bonthuis, P.J.; Geelhood, J.L.; Morlin, G.; Unrath, W.C.; Campos, J.; Crook, D.W.; Farley, M.M.; et al. Characterization of genetic and phenotypic diversity of invasive nontypeable Haemophilus influenzae. Infect. Immun. 2005, 73, 5853–5863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Pericone, C.D.; Lysenko, E.; Goldfine, H.; Weiser, J.N. Multiple mechanisms for choline transport and utilization in Haemophilus influenzae. Mol. Microbiol. 2003, 50, 537–548. [Google Scholar] [CrossRef]
- Carmody, J.M.; Herriott, R.M. Thymine and thymidine uptake by Haemophilus influenzae and the labeling of deoxyribonucleic acid. J. Bacteriol. 1970, 101, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Macfadyen, L.P.; Dorocicz, I.R.; Reizer, J.; Saier, M.H.; Redfield, R.J. Regulation of competence development and sugar utilization in Haemophilus influenzae Rd by a phosphoenolpyruvate:fructose phosphotransferase system. Mol. Microbiol. 1996, 21, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Holt, J.G.; Krieg, N.R.; Sneath, P.H.A.; Staley, J.T.; ST, W. Facultatively Anaerobic GramNegative Rods Subgroup 3: Genus Haemophilus. In Bergey’s Manual of Determinative Bacteriology, 9th ed.; Williams and Wilkins: Baltimore, MD, USA, 1994; pp. 195–280. [Google Scholar]
- Simpson, G.L.; Ortwerth, B.J. The non-oxidative degradation of ascorbic acid at physiological conditions. Biochim. Biophys. Acta 2000, 1501, 12–24. [Google Scholar] [CrossRef] [Green Version]
- Koelmel, J.; Sartain, M.; Salcedo, J.; Murali, A.; Xiangdong, L.; Stow, S. Improving Coverage of the Plasma Lipidome Using Iterative MS/MS Data Acquisition Combined with Lipid Annotator Software and 6546 LC/Q-TOF. Available online: https://www.agilent.com/cs/library/applications/application-6546-q-tof-lipidome-5994-0775en-agilent.pdf (accessed on 9 September 2020).
- Gil-de-la-Fuente, A.; Godzien, J.; Saugar, S.; Garcia-Carmona, R.; Badran, H.; Wishart, D.S.; Barbas, C.; Otero, A. CEU Mass Mediator 3.0: A Metabolite Annotation Tool. J. Proteome Res. 2019, 18, 797–802. [Google Scholar] [CrossRef]
- Han, X. Lipidomics: Comprehensive Mass Spectrometry of Lipids, 1st ed.; Wiley-Blackwell: Orlando, FL, USA, 2016. [Google Scholar]
- Plückthun, A.; Dennis, E.A. Acyl and phosphoryl migration in lysophospholipids: Importance in phospholipid synthesis and phospholipase specificity. Biochemistry 1982, 21, 1743–1750. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolite profiling in Arabidopsis. Methods Mol. Biol. 2006, 323, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Rey-Stolle, F.; Dudzik, D.; Gonzalez-Riano, C.; Fernández-García, M.; Alonso-Herranz, V.; Rojo, D.; Barbas, C.; García, A. Low and high resolution gas chromatography-mass spectrometry for untargeted metabolomics: A tutorial. Anal. Chim. Acta 2022, 1210, 339043. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, A.; Ferrarini, A.; Rey-Stolle, F.; García, A.; Barbas, C. From sample treatment to biomarker discovery: A tutorial for untargeted metabolomics based on GC-(EI)-Q-MS. Anal. Chim. Acta 2015, 900, 21–35. [Google Scholar] [CrossRef]
- López-Gonzálvez, Á.; Godzien, J.; García, A.; Barbas, C. Capillary Electrophoresis Mass Spectrometry as a Tool for Untargeted Metabolomics. Methods Mol. Biol. 2019, 1978, 55–77. [Google Scholar] [CrossRef]
- Mamani-Huanca, M.; de la Fuente, A.G.; Otero, A.; Gradillas, A.; Godzien, J.; Barbas, C.; López-Gonzálvez, Á. Enhancing confidence of metabolite annotation in Capillary Electrophoresis-Mass Spectrometry untargeted metabolomics with relative migration time and in-source fragmentation. J. Chromatogr. A 2021, 1635, 461758. [Google Scholar] [CrossRef]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- König, M.; Dräger, A.; Holzhütter, H.G. CySBML: A Cytoscape plugin for SBML. Bioinformatics 2012, 28, 2402–2403. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Utriainen, M.; Morris, J.H. clusterMaker2: A major update to clusterMaker, a multi-algorithm clustering app for Cytoscape. BMC Bioinform. 2023, 24, 134. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Zarringhalam, K.; Zhang, L.; Kiebish, M.A.; Yang, K.; Han, X.; Gross, R.W.; Chuang, J. Statistical analysis of the processes controlling choline and ethanolamine glycerophospholipid molecular species composition. PLoS ONE 2012, 7, e37293. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster Name | KOBAS-i Enriched Terms (pBH < 0.05) | Metabolite Cluster Enrichment Ratio | Cluster Enrichment pBH (Global Metabolome) | Cluster Enrichment (Correlation Islands, pBH < 0.05) |
|---|---|---|---|---|
| Cluster 1 | Pyruvate metabolism, two-component system, alanine, aspartate and glutamate metabolism, citrate cycle (TCA cycle), oxidative phosphorylation, butanoate metabolism, phosphotransferase system (PTS), amino sugar and nucleotide sugar metabolism, sulfur metabolism, biosynthesis of amino acids, methane metabolism, 2-oxocarboxylic acid metabolism, cysteine and methionine metabolism, glycolysis/gluconeogenesis, arginine biosynthesis, pantothenate and CoA biosynthesis, valine, leucine and isoleucine biosynthesis, fructose and mannose metabolism, glycerophospholipid metabolism, and nitrogen metabolism. | 1.868 | 0.094 | K, L |
| Cluster 2 | Thiamine metabolism and ABC transporters | 0 | 1.000 | - |
| Cluster 3 | Starch and sucrose metabolism, amino sugar and nucleotide sugar metabolism, galactose metabolism, and glycolysis/gluconeogenesis. | 2.163 | 1.000 | - |
| Cluster 4 | Folate biosynthesis, glyoxylate and dicarboxylate metabolism and methane metabolism. | 0.541 | 1.000 | - |
| Cluster 5 | Pentose and glucuronate interconversions, fructose and mannose metabolism, pentose phosphate pathway, ascorbate and aldarate metabolism, glycolysis/gluconeogenesis, lipopolysaccharide biosynthesis, methane metabolism, biosynthesis of amino acids, and terpenoid backbone biosynthesis. | 0.186 | 1.000 | - |
| Cluster 6 | Xanthosine and XMP transport and interconversions *. | 0 | 1.000 | - |
| Cluster 7 | Purine metabolism, biosynthesis of amino acids, pyrimidine metabolism, alanine, aspartate and glutamate metabolism, histidine metabolism, phenylalanine, tyrosine and tryptophan metabolism, vitamin B6 metabolism, 2-oxocarboxylic acid metabolism, one carbon pool by folate, glutathione metabolism, arginine biosynthesis, glycine, serine and threonine metabolism, vancomycin resistance, and nicotinate and nicotinamide metabolism | 1.287 | 0.827 | J |
| Cluster 8 | Sulfur relay system, folate biosynthesis, and ABC transporters. | 0 | 1.000 | - |
| Cluster 9 | Valine tRNA-loading * | 5.407 | 0.199 | A |
| Cluster 10 | Uridine and UMP transport and interconversions *. | 1.352 | 1.000 | D |
| Cluster 11 | Pyrimidine metabolism, purine metabolism, ABC transporters, biotin metabolism, arginine biosynthesis, and sulfur relay system. | 1.298 | 0.912 | A |
| Cluster 12 | Peptidoglycan biosynthesis, lysine metabolism, amino sugar and nucleotide sugar metabolism, biosynthesis of amino acids | 1.202 | 1.000 | - |
| Cluster 13 | Fatty acid metabolism, fatty acid biosynthesis, propanoate metabolism, pyruvate metabolism, citrate cycle (TCA cycle), lysine degradation, biotin metabolism, sulfur metabolism, cysteine and methionine metabolism, butanoate metabolism, lipopolysaccharide biosynthesis, glycerolipid metabolism, and glycerophospholipid metabolism. | 0.541 | 1.000 | - |
| Cluster 14 | Ubiquinone and other terpenoid–quinone biosynthesis, and terpenoid backbone metabolism. | 0 | 1.000 | - |
| Cluster 15 | beta-Lactam resistance, peptidoglycan biosynthesis, ABC transporters, quorum sensing, and vancomycin resistance. | 2.403 | 0.230 | - |
| Cluster 16 | Tyrosine metabolism * | 3.605 | 0.296 | E |
| Cluster 17 | Fatty acid biosynthesis, fatty acid metabolism, and quorum sensing. | 0 | 1.000 | - |
| Cluster 18 | ABC transporters and glutathione transport *. | 1.992 | 0.127 | B, F |
| Cluster 19 | Glycerophospholipid metabolism, pyrimidine metabolism, pantothenate and CoA biosynthesis, glycine, serine and threonine metabolism, lipo-polysaccharide biosynthesis, terpenoid backbone biosynthesis, phenylalanine, and tyrosine and tryptophan metabolism | 1.639 | 0.173 | - |
| Cluster 20 | One carbon pool folate, glutathione metabolism, selenocompound metabolism, and ABC transporters. | 0.94 | 1.000 | - |
| Cluster 21 | Pyrimidine metabolism, purine metabolism and nicotinate and nicotinamide metabolism. | 0 | 1.000 | - |
| Cluster 22 | Pantothenate and CoA biosynthesis, valine, leucine and isoleucine biosynthesis, biosynthesis of amino acids, arginine and proline metabolism, ABC transporters, glutathione metabolism, biosynthesis of secondary metabolism, 2-oxocarboxylic acid metabolism, C5-branched dibasic acid metabolism, butanoate metabolism, and citrate cycle (TCA cycle). | 2.028 | 0.016 | M |
| Cluster 23 | Glycine, serine and threonine metabolism, biosynthesis of amino acids, lysine biosynthesis, cysteine and methionine metabolism, vitamin B6 metabolism, and 2-oxocarboxylic acid metabolism. | 1.802 | 0.708 | M |
| Cluster 24 | Phenylalanine, tyrosine and tryptophan biosynthesis, and biosynthesis of amino acids. | 0 | 1.000 | - |
| Cluster 25 | Selenocompound metabolism and aminoacyl-tRNA biosynthesis. | 0 | 1.000 | - |
| Cluster 26 | Riboflavin metabolism, folate biosynthesis, pentose phosphate pathway, lipopolysaccharide biosynthesis, glutathione metabolism, and glyoxylate and dicarboxylate metabolism. | 0.47 | 1.000 | F |
| Cluster 27 | Nicotinate and nicotinamide metabolism, purine metabolism, and pyrimidine metabolism. | 2.704 | 0.044 | I |
| Cluster 28 | ABC transporters and ribose transport *. | 3.605 | 0.169 | I |
| Cluster 29 | Purine metabolism, nicotinate and nicotinamide metabolism, and pyrimidine metabolism. | 0 | 1.000 | - |
| Cluster 30 | ABC transporters and arginine biosynthesis. | 3.004 | 0.023 | F, K |
| Cluster 31 | Purine metabolism, nitrogen metabolism | 0 | 1.000 | - |
| Cluster 32 | ABC transporters and cationic antimicrobial peptide (CAMP) resistance. | 0 | 1.000 | - |
| Cluster 33 | Biotin metabolism, fatty acid biosynthesis, and fatty acid metabolism. | 0 | 1.000 | - |
| Cluster 34 | Aminoacyl-tRNA biosynthesis and phenylalanine transport *. | 5.407 | 0.053 | E |
| Cluster 35 | Glycine, serine and threonine metabolism, glycolysis/gluconeogenesis, methane metabolism, biosynthesis of amino acids, glyoxylate and dicarboxylate metabolism, and glycerolipid metabolism. | 2.317 | 0.184 | B |
| Cluster 36 | Glycerophospholipid metabolism, fatty acid biosynthesis, fatty acid metabolism, glycerolipid metabolism, and biotin metabolism. | 0 | 1.000 | - |
| Cluster 37 | Glycerophospholipid metabolism and glycerolipid metabolism. | 0 | 1.000 | - |
| Cluster 38 | Glycerophospholipid metabolism and glycerolipid metabolism. | 0 | 1.000 | - |
| Cluster 39 | Glycerophospholipid metabolism and glycerolipid metabolism. | 0 | 1.000 | - |
| Cluster 40 | Glycerophospholipid metabolism and glycerolipid metabolism. | 0 | 1.000 | - |
| Cluster 41 | Fatty acid biosynthesis, fatty acid metabolism, biotin metabolism, glycerophospholipid metabolism, and glycerolipid metabolism. | 0 | 1.000 | - |
| Cluster 42 | C5 branched dibasic acid metabolism, valine, leucine and isoleucine metabolism, 2-oxocarboxylic acid metabolism, biosynthesis of amino acids, and biosynthesis of secondary amino acids. | 0 | 1.000 | - |
| Cluster 43 | Thiamine metabolism | 0 | 1.000 | - |
| Cluster 44 | Pyrimidine metabolism and purine metabolism. | 1.352 | 0.675 | - |
| Cluster 45 | CMP metabolism * | 1.352 | 0.660 | - |
| Cluster 46 | dUMP metabolism * | 0 | 1.000 | - |
| Cluster 47 | Inosine metabolism * | 0 | 1.000 | - |
| Cluster 48 | ABC transporters and choline transport and incorporation into LOS *. | 2.704 | 0.174 | E |
| Cluster 49 | Leucine transport and tRNA loading *. | 5.407 | 0.037 | M |
| Cluster 50 | Histidine transport and tRNA loading. | 3.605 | 0.095 | E |
| Cluster 51 | Glycerol and glyceraldehyde metabolism *. | 1.545 | 0.397 | C |
| Cluster 52 | Biotin metabolism | 0 | 1.000 | - |
| Cluster 53 | ABC transporters and arginine transport and tRNA loading *. | 5.407 | 0.034 | E |
| Analytical Platform of Detection | Confidence in the Annotation | Matching Experimental Evidence | Metabolite Name |
|---|---|---|---|
| GC-QTOF/MS | L1 | HRMS fragmentation spectra and retention times. | Cyclo(Leu-Pro) |
| N-Methylalanine | |||
| Cadaverine | |||
| γ-Aminobutyric acid | |||
| Thymine | |||
| Cytosine | |||
| Mannose | |||
| Sucrose | |||
| Trehalose | |||
| Erythritol | |||
| Ribitol | |||
| Xylitol | |||
| Sorbitol | |||
| L2 | Nominal mass fragmentation spectra and NIST retention indices. | Pseudouridine | |
| Threonic acid | |||
| CE-TOF/MS | L1 | RMT, pseudomolecular ion m/z and in-source fragmentation. | Ophtalmic acid |
| Betaine | |||
| 5’-Methylthioadenosine |
| Fatty Acyl Chain | PE(X:0) | PG(X:0) | PE(X:1) | PG(X:1) | PE(X:2) | PG(X:2) |
|---|---|---|---|---|---|---|
| p10:0 | 0.015484 | 0.015072 | 0.007742 | 0.007536 | 0 | 0 |
| p11:0 | 0.001413 | 0.001375 | 0.000706 | 0.000688 | 0 | 0 |
| p12:0 | 0.036445 | 0.019686 | 0.018223 | 0.009843 | 0 | 0 |
| p13:0 | 0.029268 | 0.042083 | 0.014634 | 0.021042 | 0 | 0 |
| p14:0 | 0.552656 | 0.361674 | 0.276328 | 0.180837 | 0 | 0 |
| p15:0 | 0.083368 | 0.166102 | 0.041684 | 0.083051 | 0 | 0 |
| p16:0 | 0.233997 | 0.328652 | 0.116998 | 0.164326 | 0 | 0 |
| p17:0 | 0.004429 | 0.011895 | 0.002214 | 0.005948 | 0 | 0 |
| p18:0 | 0.037556 | 0.048219 | 0.018778 | 0.024109 | 0 | 0 |
| p19:0 | 0 | 0 | 0 | 0 | 0 | 0 |
| p20:0 | 0.005385 | 0.005241 | 0.002692 | 0.002621 | 0 | 0 |
| p10:1 | 0 | 0 | 0.002384 | 0.003745 | 0.004768 | 0.00749 |
| p11:1 | 0 | 0 | 0 | 0 | 0 | 0 |
| p12:1 | 0 | 0 | 0.016958 | 0.026642 | 0.033916 | 0.053284 |
| p13:1 | 0 | 0 | 0.006165 | 0.009686 | 0.01233 | 0.019371 |
| p14:1 | 0 | 0 | 0.025861 | 0.024321 | 0.051722 | 0.048642 |
| p15:1 | 0 | 0 | 0.009048 | 0.009295 | 0.018096 | 0.018589 |
| p16:1 | 0 | 0 | 0.432158 | 0.312268 | 0.864315 | 0.624535 |
| p17:1 | 0 | 0 | 0.0048 | 0.009216 | 0.0096 | 0.018432 |
| p18:1 | 0 | 0 | 0.002466 | 0.104641 | 0.004932 | 0.209282 |
| p19:1 | 0 | 0 | 0 | 0 | 0 | 0 |
| p20:1 | 0 | 0 | 0.00016 | 0.000187 | 0.00032 | 0.000374 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-García, M.; Ares-Arroyo, M.; Wedel, E.; Montero, N.; Barbas, C.; Rey-Stolle, M.F.; González-Zorn, B.; García, A. Multiplatform Metabolomics Characterization Reveals Novel Metabolites and Phospholipid Compositional Rules of Haemophilus influenzae Rd KW20. Int. J. Mol. Sci. 2023, 24, 11150. https://doi.org/10.3390/ijms241311150
Fernández-García M, Ares-Arroyo M, Wedel E, Montero N, Barbas C, Rey-Stolle MF, González-Zorn B, García A. Multiplatform Metabolomics Characterization Reveals Novel Metabolites and Phospholipid Compositional Rules of Haemophilus influenzae Rd KW20. International Journal of Molecular Sciences. 2023; 24(13):11150. https://doi.org/10.3390/ijms241311150
Chicago/Turabian StyleFernández-García, Miguel, Manuel Ares-Arroyo, Emilia Wedel, Natalia Montero, Coral Barbas, Mª Fernanda Rey-Stolle, Bruno González-Zorn, and Antonia García. 2023. "Multiplatform Metabolomics Characterization Reveals Novel Metabolites and Phospholipid Compositional Rules of Haemophilus influenzae Rd KW20" International Journal of Molecular Sciences 24, no. 13: 11150. https://doi.org/10.3390/ijms241311150
APA StyleFernández-García, M., Ares-Arroyo, M., Wedel, E., Montero, N., Barbas, C., Rey-Stolle, M. F., González-Zorn, B., & García, A. (2023). Multiplatform Metabolomics Characterization Reveals Novel Metabolites and Phospholipid Compositional Rules of Haemophilus influenzae Rd KW20. International Journal of Molecular Sciences, 24(13), 11150. https://doi.org/10.3390/ijms241311150