
DNA Methylation Signatures of Multiple Sclerosis Occur Independently of Known Genetic Risk and Are Primarily Attributed to B Cells and Monocytes
 , , , ,
, , , ,  , , , , , , ,
, , , , , , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
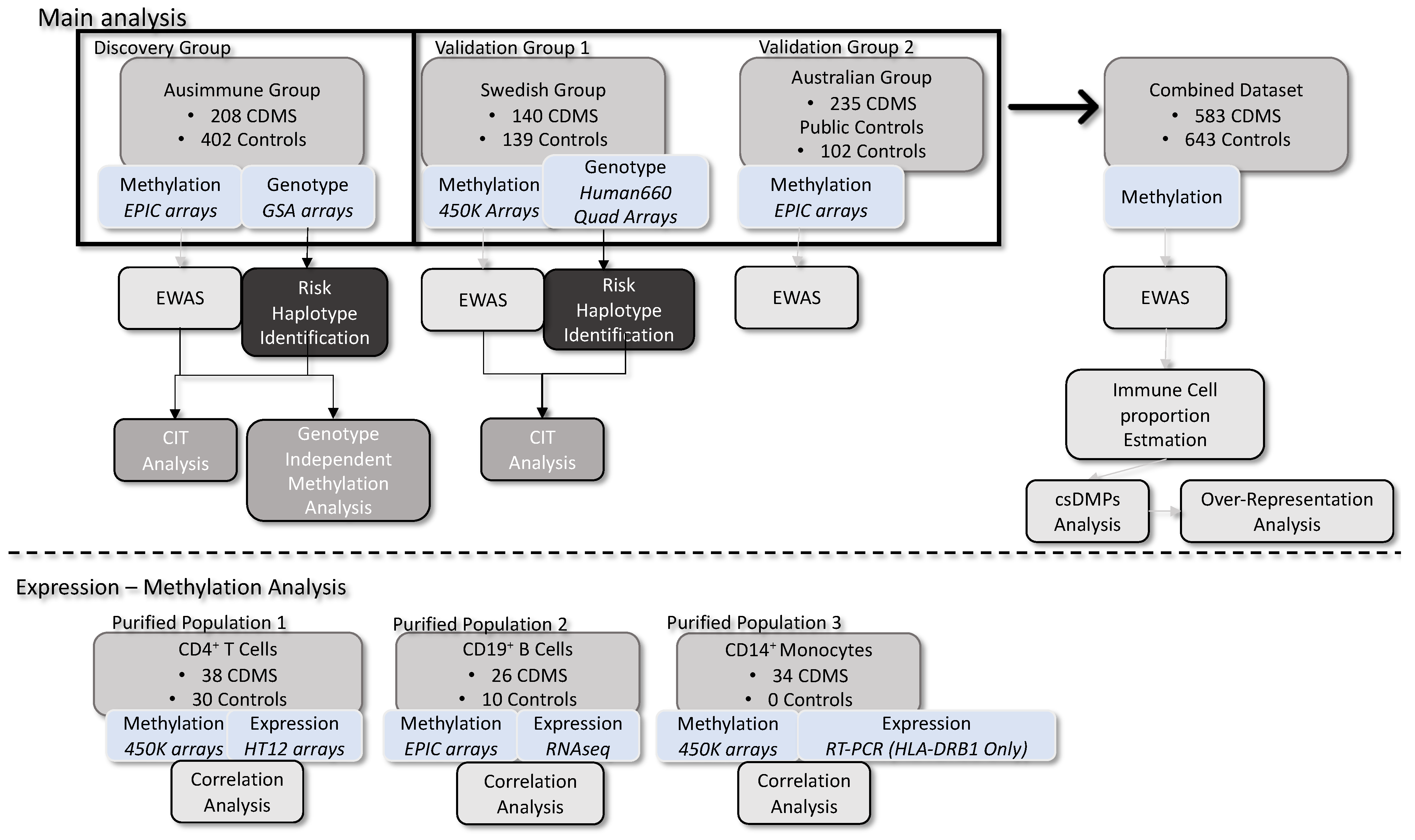
2. Results
2.1. DNA Methylation Analysis Implicates the HLA Locus
2.2. Methylation Differences Occur Independently of Known Genetic Risk
2.3. Cell-Specific Differential Methylation Is Mainly Attributed to B Cells and Monocytes
2.4. Over-Representation Analysis Shows Enrichment of DMPs in Immunological Pathways
3. Discussion
4. Materials and Methods
4.1. Whole Blood Methylation
4.2. DNA Extraction and Quality Control
4.3. Methylation and Genotyping Arrays
4.4. Methylation Analysis
4.5. Sensitivity Analysis
4.6. Genotype Analysis
4.7. Scores and Receiver Operator Characteristic Curves
4.8. Immune Cell Deconvolution Analysis
4.9. Expression and DNA Methylation Analysis
4.10. Correlation Analysis (DNA Methylation and Gene Expression)
4.11. metQTL Analysis
4.12. Over-Representation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Multiple Sclerosis Genetics Consortium; Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef] [PubMed]
- Kellar-Wood, H.F.; Wood, N.W.; Holmans, P.; Clayton, D.; Robertson, N.; Compston, D.A. Multiple sclerosis and the HLA-D region: Linkage and association studies. J. Neuroimmunol. 1995, 58, 183–190. [Google Scholar] [CrossRef]
- Sawcer, S.; Ban, M.; Maranian, M.; Yeo, T.W.; Compston, A.; Kirby, A.; Daly, M.J.; De Jager, P.L.; Walsh, E.; Lander, E.S.; et al. A high-density screen for linkage in multiple sclerosis. Am. J. Hum. Genet. 2005, 77, 454–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsopoulos, N.A.; Barcellos, L.F.; Hintzen, R.Q.; Schaefer, C.; van Duijn, C.M.; Noble, J.A.; Raj, T.; Imsgc; Anzgene; Gourraud, P.A.; et al. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet. 2013, 9, e1003926. [Google Scholar] [CrossRef]
- Attfield, K.E.; Jensen, L.T.; Kaufmann, M.; Friese, M.A.; Fugger, L. The immunology of multiple sclerosis. Nat. Rev. Immunol. 2022, 22, 734–750. [Google Scholar] [CrossRef]
- Bar-Or, A.; Li, R. Cellular immunology of relapsing multiple sclerosis: Interactions, checks, and balances. Lancet Neurol. 2021, 20, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, K. The role of Epstein-Barr virus in the etiology of multiple sclerosis: A current review. Expert. Rev. Clin. Immunol. 2020, 16, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Dunn, N.; Kharlamova, N.; Fogdell-Hahn, A. The role of herpesvirus 6A and 6B in multiple sclerosis and epilepsy. Scand. J. Immunol. 2020, 92, e12984. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, A.K. Smoking and disability progression in multiple sclerosis. Expert. Rev. Neurother. 2020, 20, 739–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, R.M.; Ponsonby, A.L.; Dear, K.; Valery, P.C.; Pender, M.P.; Taylor, B.V.; Kilpatrick, T.J.; Dwyer, T.; Coulthard, A.; Chapman, C.; et al. Sun exposure and vitamin D are independent risk factors for CNS demyelination. Neurology 2011, 76, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Ponsonby, A.L.; Lucas, R.M.; van der Mei, I.A.; Dear, K.; Valery, P.C.; Pender, M.P.; Taylor, B.V.; Kilpatrick, T.J.; Coulthard, A.; Chapman, C.; et al. Offspring number, pregnancy, and risk of a first clinical demyelinating event: The AusImmune Study. Neurology 2012, 78, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Maltby, V.E.; Graves, M.C.; Lea, R.A.; Benton, M.C.; Sanders, K.A.; Tajouri, L.; Scott, R.J.; Lechner-Scott, J. Genome-wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clin. Epigenetics 2015, 7, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltby, V.E.; Lea, R.A.; Sanders, K.A.; White, N.; Benton, M.C.; Scott, R.J.; Lechner-Scott, J. Differential methylation at MHC in CD4(+) T cells is associated with multiple sclerosis independently of HLA-DRB1. Clin. Epigenetics 2017, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Kular, L.; Liu, Y.; Ruhrmann, S.; Zheleznyakova, G.; Marabita, F.; Gomez-Cabrero, D.; James, T.; Ewing, E.; Linden, M.; Gornikiewicz, B.; et al. DNA methylation as a mediator of HLA-DRB1(star)15:01 and a protective variant in multiple sclerosis. Nat. Commun. 2018, 9, 2397. [Google Scholar] [CrossRef] [Green Version]
- Maltby, V.E.; Lea, R.A.; Graves, M.C.; Sanders, K.A.; Benton, M.C.; Tajouri, L.; Scott, R.J.; Lechner-Scott, J. Genome-wide DNA methylation changes in CD19(+) B cells from relapsing-remitting multiple sclerosis patients. Sci. Rep. 2018, 8, 17418. [Google Scholar] [CrossRef] [Green Version]
- Ewing, E.; Kular, L.; Fernandes, S.J.; Karathanasis, N.; Lagani, V.; Ruhrmann, S.; Tsamardinos, I.; Tegner, J.; Piehl, F.; Gomez-Cabrero, D.; et al. Combining evidence from four immune cell types identifies DNA methylation patterns that implicate functionally distinct pathways during Multiple Sclerosis progression. eBioMedicine 2019, 43, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Bos, S.D.; Page, C.M.; Andreassen, B.K.; Elboudwarej, E.; Gustavsen, M.W.; Briggs, F.; Quach, H.; Leikfoss, I.S.; Bjolgerud, A.; Berge, T.; et al. Genome-wide DNA methylation profiles indicate CD8+ T cell hypermethylation in multiple sclerosis. PLoS ONE 2015, 10, e0117403. [Google Scholar] [CrossRef] [Green Version]
- Rhead, B.; Brorson, I.S.; Berge, T.; Adams, C.; Quach, H.; Moen, S.M.; Berg-Hansen, P.; Celius, E.G.; Sangurdekar, D.P.; Bronson, P.G.; et al. Increased DNA methylation of SLFN12 in CD4+ and CD8+ T cells from multiple sclerosis patients. PLoS ONE 2018, 13, e0206511. [Google Scholar] [CrossRef] [Green Version]
- Campagna, M.P.; Xavier, A.; Lechner-Scott, J.; Maltby, V.; Scott, R.J.; Butzkueven, H.; Jokubaitis, V.G.; Lea, R.A. Epigenome-wide association studies: Current knowledge, strategies and recommendations. Clin. Epigenetics 2021, 13, 214. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Ponsonby, A.L.; McMichael, A.; van der Mei, I.; Chapman, C.; Coulthard, A.; Dear, K.; Dwyer, T.; Kilpatrick, T.; Pender, M.; et al. Observational analytic studies in multiple sclerosis: Controlling bias through study design and conduct. The Australian Multicentre Study of Environment and Immune Function. Mult. Scler. 2007, 13, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Millstein, J.; Zhang, B.; Zhu, J.; Schadt, E.E. Disentangling molecular relationships with a causal inference test. BMC Genet. 2009, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.W.; O’Reilly, P.F. PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience 2019, 8, giz082. [Google Scholar] [CrossRef]
- Zheng, S.C.; Breeze, C.E.; Beck, S.; Teschendorff, A.E. Identification of differentially methylated cell types in epigenome-wide association studies. Nat. Methods 2018, 15, 1059–1066. [Google Scholar] [CrossRef]
- Ong, L.T.C.; Schibeci, S.D.; Fewings, N.L.; Booth, D.R.; Parnell, G.P. Age-dependent VDR peak DNA methylation as a mechanism for latitude-dependent multiple sclerosis risk. Epigenetics Chromatin 2021, 14, 9. [Google Scholar] [CrossRef]
- Marabita, F.; Almgren, M.; Sjoholm, L.K.; Kular, L.; Liu, Y.; James, T.; Kiss, N.B.; Feinberg, A.P.; Olsson, T.; Kockum, I.; et al. Smoking induces DNA methylation changes in Multiple Sclerosis patients with exposure-response relationship. Sci. Rep. 2017, 7, 14589. [Google Scholar] [CrossRef] [Green Version]
- Christopoulos, P.F.; Gjolberg, T.T.; Kruger, S.; Haraldsen, G.; Andersen, J.T.; Sundlisaeter, E. Targeting the Notch Signaling Pathway in Chronic Inflammatory Diseases. Front. Immunol. 2021, 12, 668207. [Google Scholar] [CrossRef]
- Lee, W.S.; Lee, W.H.; Bae, Y.C.; Suk, K. Axon Guidance Molecules Guiding Neuroinflammation. Exp. Neurobiol. 2019, 28, 311–319. [Google Scholar] [CrossRef]
- Dominguez-Romero, M.E.; Slater, P.G. Unraveling Axon Guidance during Axotomy and Regeneration. Int. J. Mol. Sci. 2021, 22, 8344. [Google Scholar] [CrossRef]
- Wang, S.Q.; Wang, Y.X.; Zou, S.Q. A Glance at the Molecules That Regulate Oligodendrocyte Myelination. Curr. Issues Mol. Biol. 2022, 44, 2194–2216. [Google Scholar] [CrossRef] [PubMed]
- Graves, M.; Benton, M.; Lea, R.; Boyle, M.; Tajouri, L.; Macartney-Coxson, D.; Scott, R.; Lechner-Scott, J. Methylation differences at the HLA-DRB1 locus in CD4+ T-Cells are associated with multiple sclerosis. Mult. Scler. 2013, 20, 1033–1041. [Google Scholar] [CrossRef]
- Ma, Q.; Caillier, S.J.; Muzic, S.; University of California San Francisco MS-EPIC Team; Wilson, M.R.; Henry, R.G.; Cree, B.A.C.; Hauser, S.L.; Didonna, A.; Oksenberg, J.R. Specific hypomethylation programs underpin B cell activation in early multiple sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2111920118. [Google Scholar] [CrossRef]
- Sabatino, J.J., Jr.; Zamvil, S.S.; Hauser, S.L. B-Cell Therapies in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a032037. [Google Scholar] [CrossRef] [Green Version]
- Bittner, S.; Ruck, T.; Wiendl, H.; Grauer, O.M.; Meuth, S.G. Targeting B cells in relapsing-remitting multiple sclerosis: From pathophysiology to optimal clinical management. Ther. Adv. Neurol. Disord. 2017, 10, 51–66. [Google Scholar] [CrossRef]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef] [Green Version]
- Bar-Or, A.; Nuttall, R.K.; Duddy, M.; Alter, A.; Kim, H.J.; Ifergan, I.; Pennington, C.J.; Bourgoin, P.; Edwards, D.R.; Yong, V.W. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain 2003, 126, 2738–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, E.; Zanghi, A.; Parrinello, N.L.; Romano, A.; Palumbo, G.A.; Chisari, C.G.; Toscano, S.; Raimondo, F.D.; Zappia, M.; Patti, F. Immunological Subsets Characterization in Newly Diagnosed Relapsing-Remitting Multiple Sclerosis. Front. Immunol. 2022, 13, 819136. [Google Scholar] [CrossRef] [PubMed]
- Durelli, L.; Conti, L.; Clerico, M.; Boselli, D.; Contessa, G.; Ripellino, P.; Ferrero, B.; Eid, P.; Novelli, F. T-Helper 17 Cells Expand in Multiple Sclerosis and Are Inhibited by Interferon-beta. Ann. Neurol. 2009, 65, 499–509. [Google Scholar] [CrossRef]
- Zhang, X.; Tao, Y.Z.; Chopra, M.; Dujmovic-Basuroski, I.; Jin, J.P.; Tang, Y.N.; Drulovic, J.; Markovic-Pese, S. IL-11 Induces Th17 Cell Responses in Patients with Early Relapsing-Remitting Multiple Sclerosis. J. Immunol. 2015, 194, 5139–5149. [Google Scholar] [CrossRef] [Green Version]
- Pender, M.P.; Csurhes, P.A.; Pfluger, C.M.M.; Burrows, S.R. Deficiency of CD8(+) effector memory T cells is an early and persistent feature of multiple sclerosis. Mult. Scler. J. 2014, 20, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Caruana, P.; Lemmert, K.; Ribbons, K.; Lea, R.; Lechner-Scott, J. Natural killer cell subpopulations are associated with MRI activity in a relapsing-remitting multiple sclerosis patient cohort from Australia. Mult. Scler. J. 2017, 23, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [Green Version]
- Salgado, F.J.; Lojo, J.; Fernandez-Alonso, C.M.; Vinuela, J.; Cordero, O.J.; Nogueira, M. Interleukin-dependent modulation of HLA-DR expression on CD4and CD8 activated T cells. Immunol. Cell Biol. 2002, 80, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Diniz, S.N.; da Silva, C.F.; de Almeida, I.T.; da Silva Costa, F.E.; de Oliveira, E.M.L. INFbeta treatment affects global DNA methylation in monocytes of patients with multiple sclerosis. J. Neuroimmunol. 2021, 355, 577563. [Google Scholar] [CrossRef] [PubMed]
- Souren, N.Y.; Gerdes, L.A.; Lutsik, P.; Gasparoni, G.; Beltran, E.; Salhab, A.; Kumpfel, T.; Weichenhan, D.; Plass, C.; Hohlfeld, R.; et al. DNA methylation signatures of monozygotic twins clinically discordant for multiple sclerosis. Nat. Commun. 2019, 10, 2094. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2017, 17, 162–173. [Google Scholar] [CrossRef]
- Campagna, M.P.; Xavier, A.; Lea, R.A.; Stankovich, J.; Maltby, V.E.; Butzkueven, H.; Lechner-Scott, J.; Scott, R.J.; Jokubaitis, V.G. Whole-blood methylation signatures are associated with and accurately classify multiple sclerosis disease severity. Clin. Epigenetics 2022, 14, 194. [Google Scholar] [CrossRef]
- Yan, L.; Ma, C.; Wang, D.; Hu, Q.; Qin, M.; Conroy, J.M.; Sucheston, L.E.; Ambrosone, C.B.; Johnson, C.S.; Wang, J.; et al. OSAT: A tool for sample-to-batch allocations in genomics experiments. BMC Genom. 2012, 13, 689. [Google Scholar] [CrossRef] [Green Version]
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2014, 30, 428–430. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef] [Green Version]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; He, J.; Zhao, S.; Wu, H.; Zhong, X.; Sheng, Q.; Samuels, D.C.; Shyr, Y.; Long, J. Illumina human exome genotyping array clustering and quality control. Nat. Protoc. 2014, 9, 2643–2662. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zivkovic, M.; Stankovic, A.; Dincic, E.; Popovic, M.; Popovic, S.; Raicevic, R.; Alavantic, D. The tag SNP for HLA-DRB1*1501, rs3135388, is significantly associated with multiple sclerosis susceptibility: Cost-effective high-throughput detection by real-time PCR. Clin. Chim. Acta 2009, 406, 27–30. [Google Scholar] [CrossRef]
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. ROCR: Visualizing classifier performance in R. Bioinformatics 2005, 21, 3940–3941. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Breeze, C.E.; Zheng, S.J.C.; Beck, S. A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinform. 2017, 18, 105. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Dunning, M.J.; Smith, M.L.; Ritchie, M.E.; Tavare, S. beadarray: R classes and methods for Illumina bead-based data. Bioinformatics 2007, 23, 2183–2184. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Marabita, F.; Almgren, M.; Lindholm, M.E.; Ruhrmann, S.; Fagerstrom-Billai, F.; Jagodic, M.; Sundberg, C.J.; Ekstrom, T.J.; Teschendorff, A.E.; Tegner, J.; et al. An evaluation of analysis pipelines for DNA methylation profiling using the Illumina HumanMethylation450 BeadChip platform. Epigenetics 2013, 8, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; CC, Y.W.; Volta, M.; Lunnon, K.; Mill, J.; Schalkwyk, L.C. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genom. 2013, 14, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2013, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xavier, A.; Maltby, V.E.; Ewing, E.; Campagna, M.P.; Burnard, S.M.; Tegner, J.N.; Slee, M.; Butzkueven, H.; Kockum, I.; Kular, L.; et al. DNA Methylation Signatures of Multiple Sclerosis Occur Independently of Known Genetic Risk and Are Primarily Attributed to B Cells and Monocytes. Int. J. Mol. Sci. 2023, 24, 12576. https://doi.org/10.3390/ijms241612576
Xavier A, Maltby VE, Ewing E, Campagna MP, Burnard SM, Tegner JN, Slee M, Butzkueven H, Kockum I, Kular L, et al. DNA Methylation Signatures of Multiple Sclerosis Occur Independently of Known Genetic Risk and Are Primarily Attributed to B Cells and Monocytes. International Journal of Molecular Sciences. 2023; 24(16):12576. https://doi.org/10.3390/ijms241612576
Chicago/Turabian StyleXavier, Alexandre, Vicki E. Maltby, Ewoud Ewing, Maria Pia Campagna, Sean M. Burnard, Jesper N. Tegner, Mark Slee, Helmut Butzkueven, Ingrid Kockum, Lara Kular, and et al. 2023. "DNA Methylation Signatures of Multiple Sclerosis Occur Independently of Known Genetic Risk and Are Primarily Attributed to B Cells and Monocytes" International Journal of Molecular Sciences 24, no. 16: 12576. https://doi.org/10.3390/ijms241612576
APA StyleXavier, A., Maltby, V. E., Ewing, E., Campagna, M. P., Burnard, S. M., Tegner, J. N., Slee, M., Butzkueven, H., Kockum, I., Kular, L., Ausimmune/AusLong Investigators Group, Jokubaitis, V. G., Kilpatrick, T., Alfredsson, L., Jagodic, M., Ponsonby, A. -L., Taylor, B. V., Scott, R. J., Lea, R. A., & Lechner-Scott, J. (2023). DNA Methylation Signatures of Multiple Sclerosis Occur Independently of Known Genetic Risk and Are Primarily Attributed to B Cells and Monocytes. International Journal of Molecular Sciences, 24(16), 12576. https://doi.org/10.3390/ijms241612576