
DMSO-Induced Unfolding of the Antifungal Disulfide Protein PAF and Its Inactive Variant: A Combined NMR and DSC Study

 and
and 
Abstract
:1. Introduction
2. Results
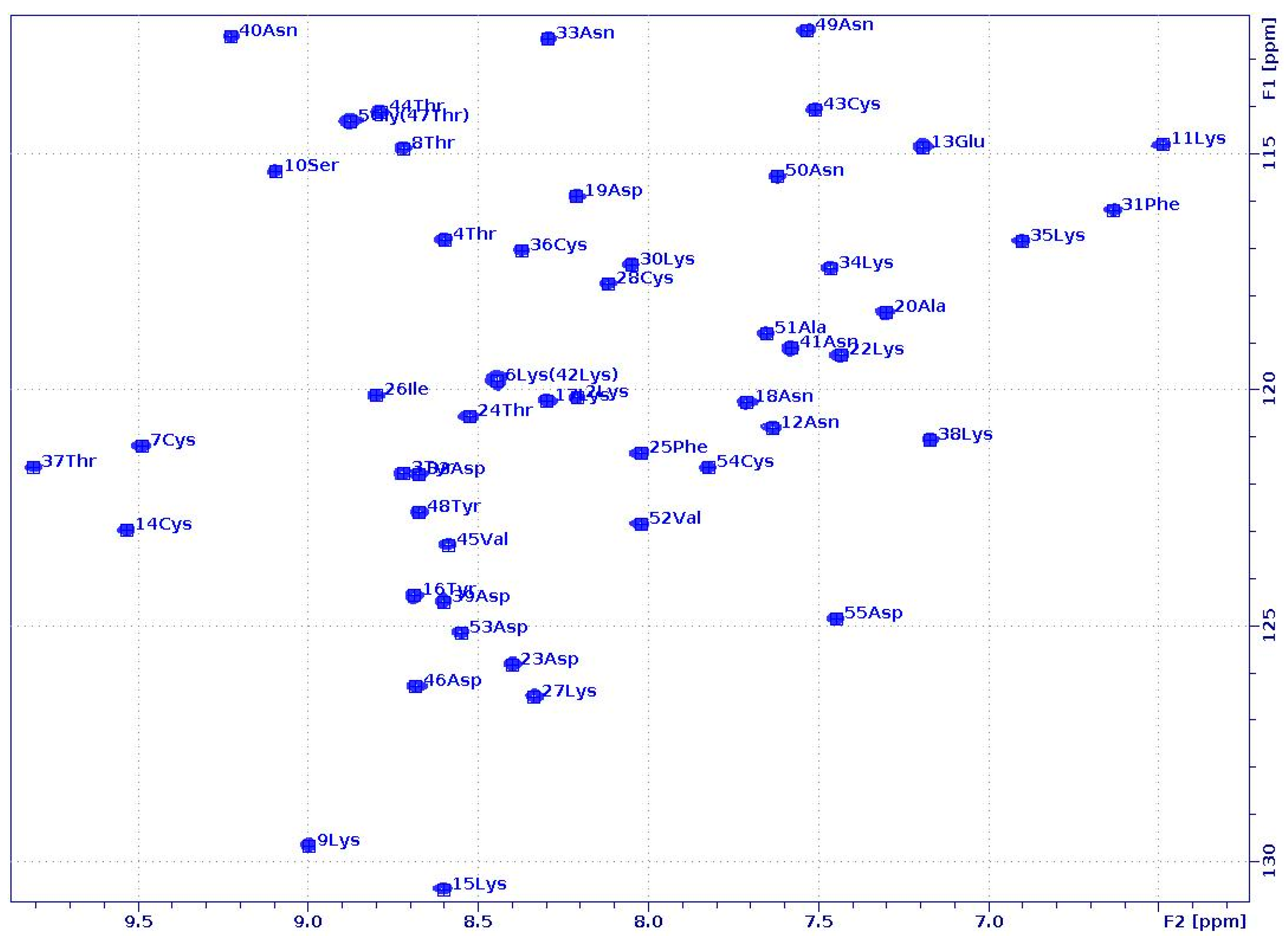
2.1. PAF and PAFD19S Have Similar Solution Structures in Water and in a 50% DMSO–Water Mixture
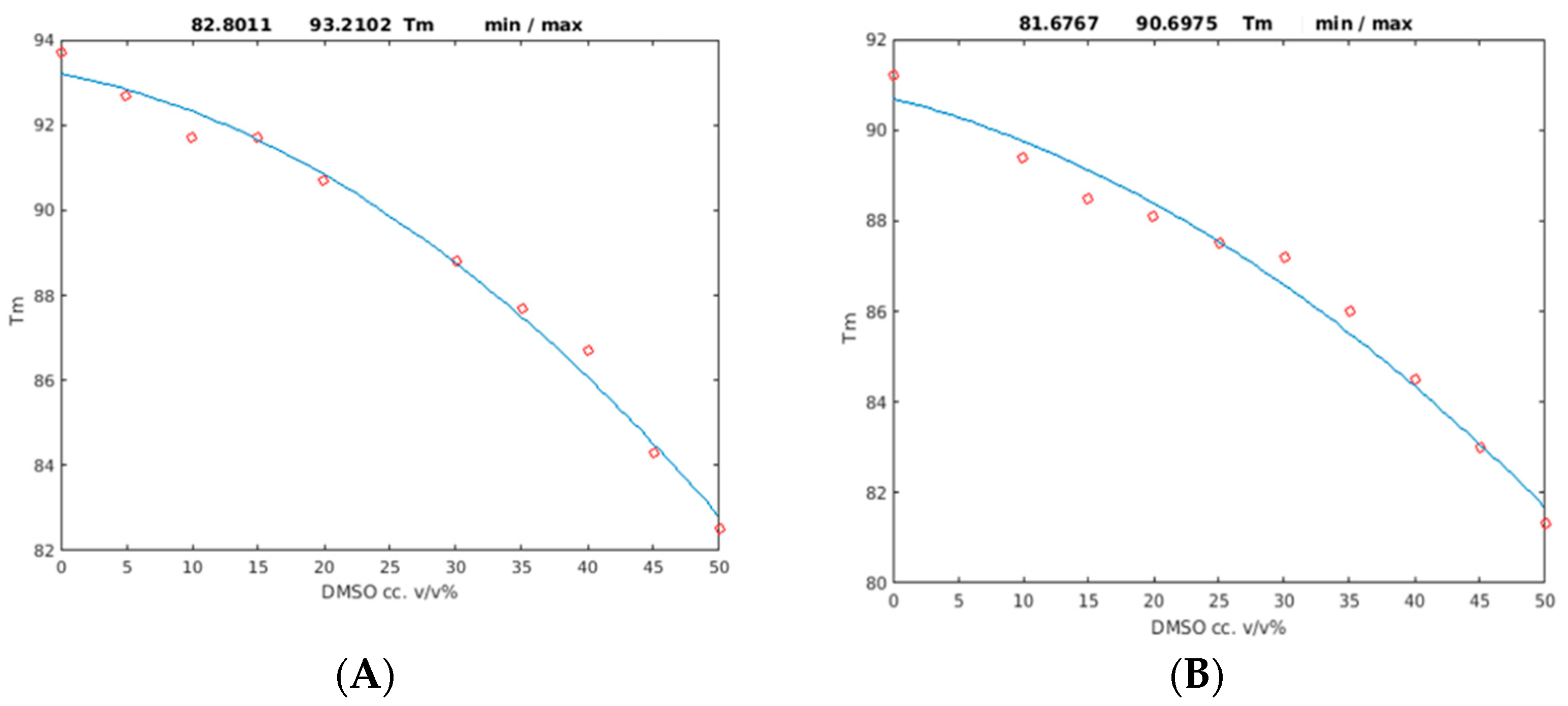
2.2. Unfolding Monitored by Differential Scanning Calorimetry (DSC)
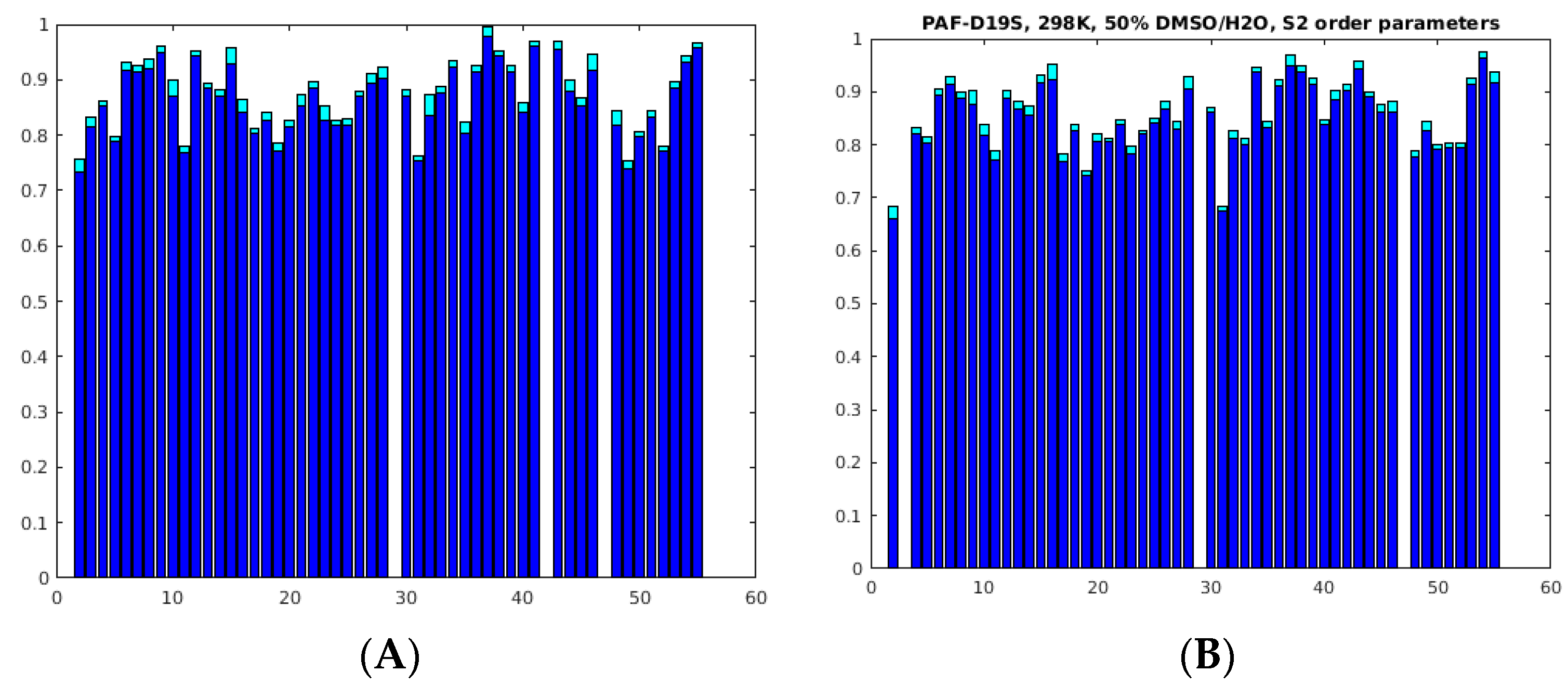
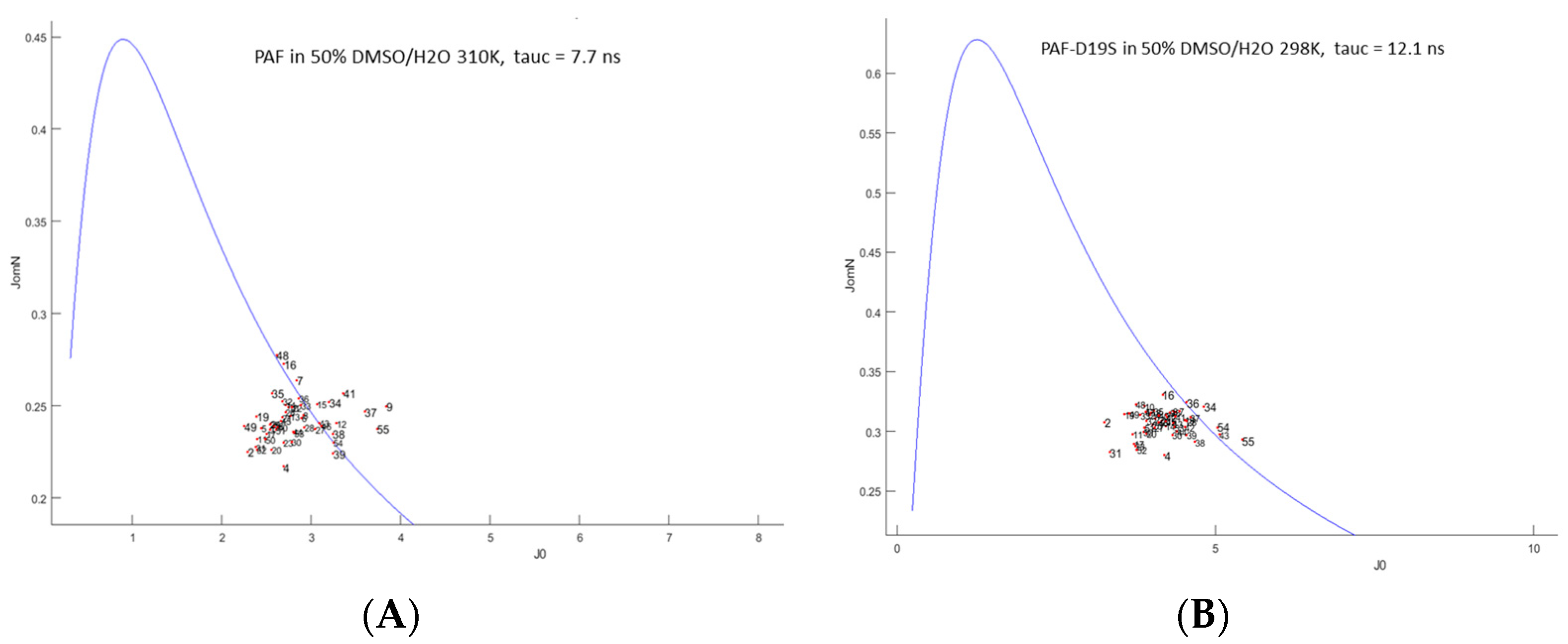
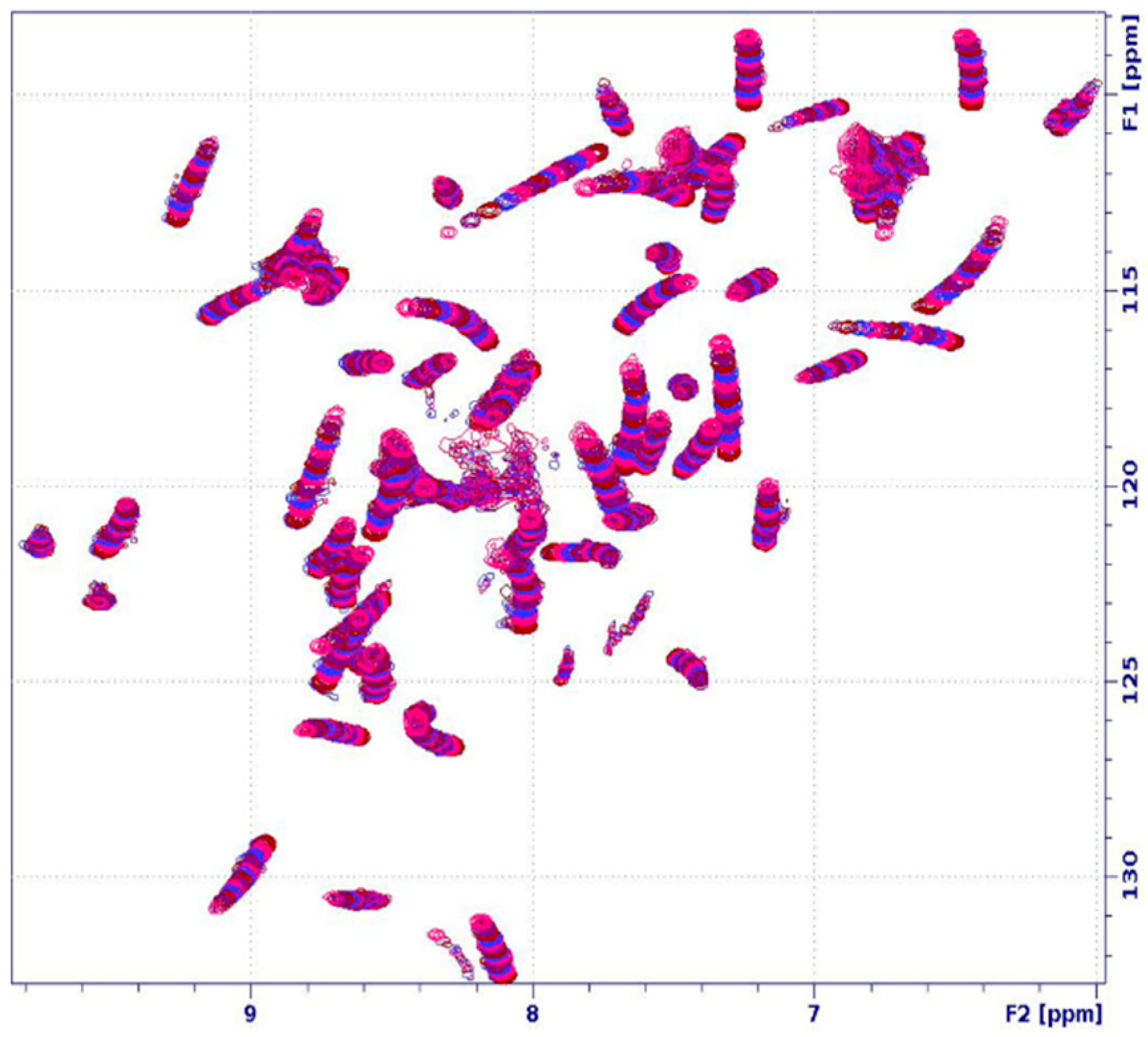
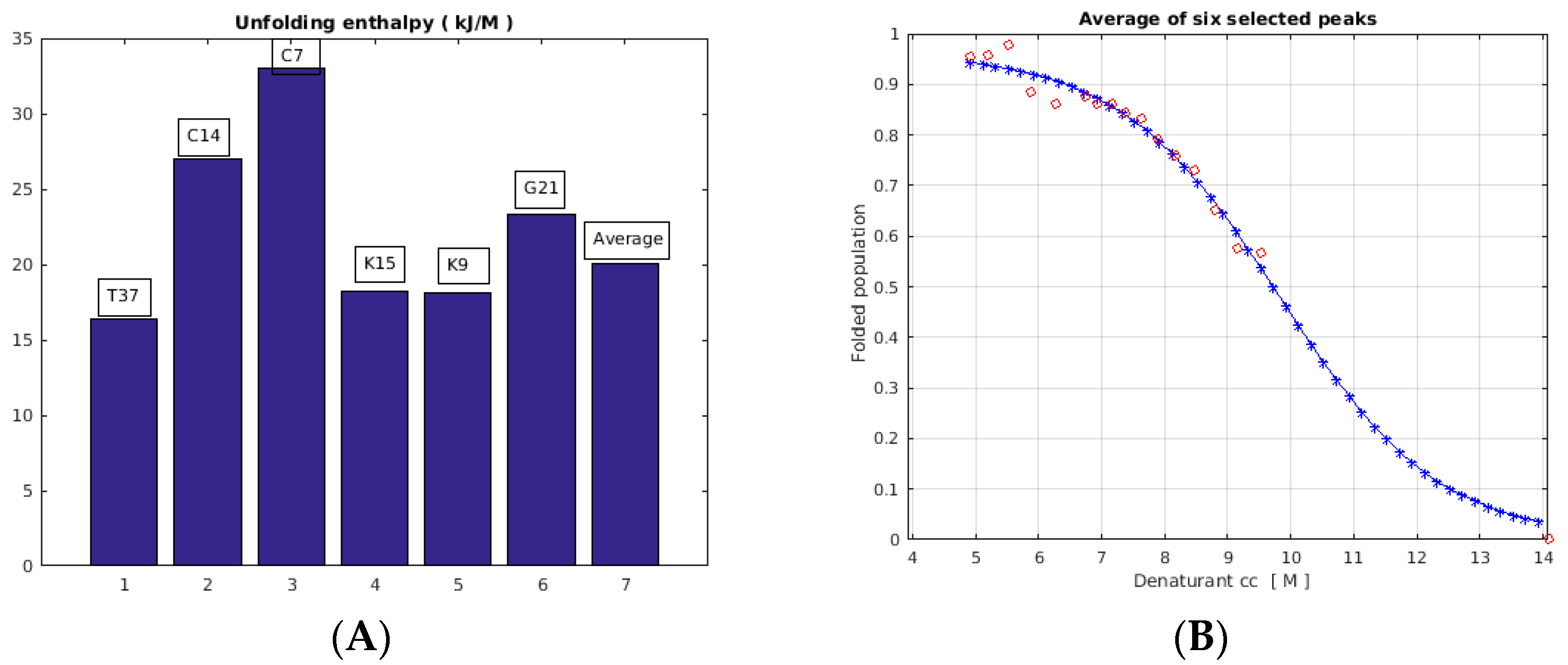
2.3. Unfolding Monitored by NMR

3. Discussion
4. Materials and Methods
4.1. Differential Scanning Calorimetry (DSC)
4.2. NMR Spectroscopy: Signal Assignments, Structure Calculations, and DMSO Titration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden Killers: Human Fungal Infections. Sci. Transl. Med. 2012, 4, 165. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Brown, K.L.; Hancock, R.E.W. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- van der Weerden, N.L.; Bleackley, M.R.; Anderson, M.A. Properties and mechanisms of action of naturally occurring antifungal peptides. Cell. Mol. Life Sci. 2013, 70, 3545–3570. [Google Scholar] [CrossRef]
- Neelabh; Singh, K.; Rani, J. Sequential and Structural Aspects of Antifungal Peptides from Animals, Bacteria and Fungi Based on Bioinformatics Tools. Probiotics Antimicrob. Proteins 2016, 8, 85–101. [Google Scholar] [CrossRef]
- Galgoczy, L.; Yap, A.; Marx, F. Cysteine-Rich Antifungal Proteins from Filamentous Fungi are Promising Bioactive Natural Compounds in Anti-Candida Therapy. Isr. J. Chem. 2019, 59, 360–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, F.; Binder, U.; Leiter, E.; Pocsi, I. The Penicillium chrysogenum antifungal protein PAF, a promising tool for the development of new antifungal therapies and fungal cell biology studies. Cell. Mol. Life Sci. 2008, 65, 445–454. [Google Scholar] [CrossRef]
- Meyer, V. A small protein that fights fungi: AFP as a new promising antifungal agent of biotechnological value. Appl. Microbiol. Biotechnol. 2008, 78, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Cools, T.L.; Struyfs, C.; Cammue, B.P.A.; Thevissen, K. Antifungal plant defensins: Increased insight in their mode of action as a basis for their use to combat fungal infections. Future Microbiol. 2017, 12, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Galgoczy, L.; Viragh, M.; Kovacs, L.; Toth, B.; Papp, T.; Vagvoelgyi, C. Antifungal peptides homologous to the Penicillium chrysogenum antifungal protein (PAF) are widespread among Fusaria. Peptides 2013, 39, 131–137. [Google Scholar] [CrossRef]
- Szappanos, H.; Szigeti, G.P.; Pal, B.; Rusznak, Z.; Szucs, G.; Rajnavolgyi, E.; Balla, J.; Balla, G.; Nagy, E.; Leiter, E.; et al. The antifungal protein AFP secreted by Aspergillus giganteus does not cause detrimental effects on certain mammalian cells. Peptides 2006, 27, 1717–1725. [Google Scholar] [CrossRef]
- Kovacs, R.; Holzknecht, J.; Hargital, Z.; Papp, C.; Farkas, A.; Borics, A.; Toth, L.; Varadi, G.; Toth, G.K.; Kovacs, I.; et al. In Vivo Applicability of Neosartorya fischeri Antifungal Protein 2 (NFAP2) in Treatment of Vulvovaginal Candidiasis. Antimicrob. Agents Chemother. 2019, 63, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palicz, Z.; Jenes, A.; Gall, T.; Miszti-Blasius, K.; Kollar, S.; Kovacs, I.; Emri, M.; Marian, T.; Leiter, E.; Pocsi, I.; et al. In vivo application of a small molecular weight antifungal protein of Penicillium chrysogenum (PAF). Toxicol. Appl. Pharmacol. 2013, 269, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Camposolivas, R.; Bruix, M.; Santoro, J.; Lacadena, J.; Delpozo, A.M.; Gavilanes, J.G.; Rico, M. Nmr solution structure of the antifungal protein from aspergillus-giganteus-evidence for cysteine pairing isomerism. Biochemistry 1995, 34, 3009–3021. [Google Scholar] [CrossRef] [PubMed]
- Batta, G.; Barna, T.; Gaspari, Z.; Sandor, S.; Koever, K.E.; Binder, U.; Sarg, B.; Kaiserer, L.; Chhillar, A.K.; Eigentler, A.; et al. Functional aspects of the solution structure and dynamics of PAF—A highly-stable antifungal protein from Penicillium chrysogenum. FEBS J. 2009, 276, 2875–2890. [Google Scholar] [CrossRef] [Green Version]
- Alex, J.M.; Rennie, M.L.; Engilberge, S.; Lehoczki, G.; Dorottya, H.; Fizil, A.; Batta, G.; Crowley, P.B. Calixarene-mediated assembly of a small antifungal protein. Iucrj 2019, 6, 238–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, L.; Viragh, M.; Tako, M.; Papp, T.; Vagvoelgyi, C.; Galgoczy, L. Isolation and characterization of Neosartorya fischeri antifungal protein (NFAP). Peptides 2011, 32, 1724–1731. [Google Scholar] [CrossRef]
- Toth, L.; Kele, Z.; Borics, A.; Nagy, L.G.; Varadi, G.; Viragh, M.; Tako, M.; Vagvolgyi, C.; Galgoczy, L. NFAP2, a novel cysteine-rich anti-yeast protein from Neosartorya fischeri NRRL 181: Isolation and characterization. Amb Express 2016, 6, 75. [Google Scholar] [CrossRef] [Green Version]
- Toth, L.; Varadi, G.; Borics, A.; Batta, G.; Kele, Z.; Vendrinszky, A.; Toth, R.; Ficze, H.; Toth, G.K.; Vagyolgyi, C.; et al. Anti-Candidal Activity and Functional Mapping of Recombinant and Synthetic Neosartorya fischeri Antifungal Protein 2 (NFAP2). Front. Microbiol. 2018, 9, 393. [Google Scholar] [CrossRef]
- Viragh, M.; Marton, A.; Vizler, C.; Toth, L.; Vagvoelgyi, C.; Marx, F.; Galgoczy, L. Insight into the antifungal mechanism of Neosartorya fischeri antifungal protein. Protein Cell 2015, 6, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Galgoczy, L.; Borics, A.; Viragh, M.; Ficze, H.; Varadi, G.; Kele, Z.; Marx, F. Structural determinants of Neosartorya fischeri antifungal protein (NFAP) for folding, stability and antifungal activity. Sci. Rep. 2017, 7, 1963. [Google Scholar] [CrossRef] [Green Version]
- Hajdu, D.; Huber, A.; Czajlik, A.; Toth, L.; Kele, Z.; Kocsube, S.; Fizil, A.; Marx, F.; Galgoczy, L.; Batta, G. Solution structure and novel insights into phylogeny and mode of action of the Neosartorya (Aspergillus) fischeri antifungal protein (NFAP). Int. J. Biol. Macromol. 2019, 129, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Leiter, E.; Szappanos, H.; Oberparleiter, C.; Kaiserer, L.; Csernoch, L.; Pusztahelyi, T.; Emri, T.; Pocsi, I.; Salvenmoser, W.; Marx, F. Antifungal protein PAF severely affects the integrity of the plasma membrane of Aspergillus nidulans and induces an apoptosis-like phenotype. Antimicrob. Agents Chemother. 2005, 49, 2445–2453. [Google Scholar] [CrossRef] [Green Version]
- Huber, A.; Galgoczy, L.; Varadi, G.; Holzknecht, J.; Kakar, A.; Malanovic, N.; Leber, R.; Koch, J.; Keller, M.A.; Batta, G.; et al. Two small, cysteine-rich and cationic antifungal proteins from Penicillium chrysogenum: A comparative study of PAF and PAFB. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 8. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, N.; Leiter, E.; Kovacs, B.; Tomori, V.; Kwon, N.-J.; Emri, T.; Marx, F.; Batta, G.; Csernoch, L.; Haas, H.; et al. The small molecular mass antifungal protein of Penicillium chrysogenum—A mechanism of action oriented review. J. Basic Microbiol. 2011, 51, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Binder, U.; Bencina, M.; Fizil, A.; Batta, G.; Chhillar, A.K.; Marx, F. Protein kinase A signaling and calcium ions are major players in PAF mediated toxicity against Aspergillus niger. FEBS Lett. 2015, 589, 1266–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fizil, A.; Gaspari, Z.; Barna, T.; Marx, F.; Batta, G. “Invisible” Conformers of an Antifungal Disulfide Protein Revealed by Constrained Cold and Heat Unfolding, CEST-NMR Experiments, and Molecular Dynamics Calculations. Chem.-A Eur. J. 2015, 21, 5136–5144. [Google Scholar] [CrossRef] [Green Version]
- Sonderegger, C.; Fizil, A.; Burtscher, L.; Hajdu, D.; Munoz, A.; Gaspari, Z.; Read, N.D.; Batta, G.; Marx, F. D19S Mutation of the Cationic, Cysteine-Rich Protein PAF: Novel Insights into Its Structural Dynamics, Thermal Unfolding and Antifungal Function. PLoS ONE 2017, 12, e0169920. [Google Scholar] [CrossRef] [Green Version]
- Varadi, G.; Toth, G.K.; Batta, G. Structure and Synthesis of Antifungal Disulfide beta-Strand Proteins from Filamentous Fungi. Microorganisms 2019, 7, 5. [Google Scholar] [CrossRef]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Formankay, J.D.; Kay, L.E. Backbone dynamics of a free and a phosphopeptide-complexed src homology-2 domain studied by n-15 nmr relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 1. theory and range of validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Lefevre, J.F.; Dayie, K.T.; Peng, J.W.; Wagner, G. Internal mobility in the partially folded DNA binding and dimerization domains of GAL4: NMR analysis of the N-H spectral density functions. Biochemistry 1996, 35, 2674–2686. [Google Scholar] [CrossRef] [PubMed]
- Kneller, J.M.; Lu, M.; Bracken, C. An effective method for the discrimination of motional anisotropy and chemical exchange. J. Am. Chem. Soc. 2002, 124, 1852–1853. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.H.; Xi, W.H.; Nussinov, R.; Ma, B.Y. Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem. Rev. 2016, 116, 6516–6551. [Google Scholar] [CrossRef]
- Vallurupalli, P.; Bouvignies, G.; Kay, L.E. Studying “Invisible” Excited Protein States in Slow Exchange with a Major State Conformation. J. Am. Chem. Soc. 2012, 134, 8148–8161. [Google Scholar] [CrossRef]
- Julien, O.; Chatterjee, S.; Bjorndahl, T.C.; Sweeting, B.; Acharya, S.; Semenchenko, V.; Chakrabartty, A.; Pai, E.F.; Wishart, D.S.; Sykes, B.D.; et al. Relative and Regional Stabilities of the Hamster, Mouse, Rabbit, and Bovine Prion Proteins toward Urea Unfolding Assessed by Nuclear Magnetic Resonance and Circular Dichroism Spectroscopies. Biochemistry 2011, 50, 7536–7545. [Google Scholar] [CrossRef]
- Huber, A.; Hajdu, D.; Bratschun-Khan, D.; Gaspari, Z.; Varbanov, M.; Philippot, S.; Fizil, A.; Czajlik, A.; Kele, Z.; Sonderegger, C.; et al. New Antimicrobial Potential and Structural Properties of PAFB: A Cationic, Cysteine-Rich Protein from Penicillium chrysogenum Q176. Sci. Rep. 2018, 8, 1751. [Google Scholar] [CrossRef] [Green Version]
- Voets, I.K.; Cruz, W.A.; Moitzi, C.; Lindner, P.; Areas, E.P.G.; Schurtenberger, P. DMSO-Induced Denaturation of Hen Egg White Lysozyme. J. Phys. Chem. B 2010, 114, 11875–11883. [Google Scholar] [CrossRef]
- Sonderegger, C.; Galgoczy, L.; Garrigues, S.; Fizil, A.; Borics, A.; Manzanares, P.; Hegedues, N.; Huber, A.; Marcos, J.F.; Batta, G.; et al. A Penicillium chrysogenum-based expression system for the production of small, cysteine-rich antifungal proteins for structural and functional analyses. Microb. Cell Factories 2016, 15, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, S.P.; Fogh, R.H.; Boucher, W.; Ragan, T.J.; Mureddu, L.G.; Vuister, G.W. CcpNmr AnalysisAssign: A flexible platform for integrated NMR analysis. J. Biomol. Nmr 2016, 66, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, P.; Pedrini, B.; Mohanty, B.; Geralt, M.; Herrmann, T.; Wuthrich, K. The J-UNIO protocol for automated protein structure determination by NMR in solution. J. Biomol. Nmr 2012, 53, 341–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guntert, P.; Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. Nmr 2015, 62, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Abeygunawardana, C.; Johnson, M.O.; Vanzijl, P.C.M. Improved sensitivity of hsqc spectra of exchanging protons at short interscan delays using a new fast hsqc (fhsqc) detection scheme that avoids water saturation. J. Magn. Reson. Ser. B 1995, 108, 94–98. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | p1 (* 10−3) | p2 (* 10−3) | Tm (°C) (p3) (0% DMSO) | Tm (°C) (50% DMSO) | R2 |
|---|---|---|---|---|---|
| PAF | −2.98 | −59.0 | 93.2 ± 0.8 | 82.8 ± 0.7 | 0.99 |
| PAFD19S | −2.16 | −72.2 | 90.7 ± 1.0 | 81.7 ± 0.9 | 0.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czajlik, A.; Batta, Á.; Kerner, K.; Fizil, Á.; Hajdu, D.; Raics, M.; Kövér, K.E.; Batta, G. DMSO-Induced Unfolding of the Antifungal Disulfide Protein PAF and Its Inactive Variant: A Combined NMR and DSC Study. Int. J. Mol. Sci. 2023, 24, 1208. https://doi.org/10.3390/ijms24021208
Czajlik A, Batta Á, Kerner K, Fizil Á, Hajdu D, Raics M, Kövér KE, Batta G. DMSO-Induced Unfolding of the Antifungal Disulfide Protein PAF and Its Inactive Variant: A Combined NMR and DSC Study. International Journal of Molecular Sciences. 2023; 24(2):1208. https://doi.org/10.3390/ijms24021208
Chicago/Turabian StyleCzajlik, András, Ágnes Batta, Kinga Kerner, Ádám Fizil, Dorottya Hajdu, Mária Raics, Katalin E. Kövér, and Gyula Batta. 2023. "DMSO-Induced Unfolding of the Antifungal Disulfide Protein PAF and Its Inactive Variant: A Combined NMR and DSC Study" International Journal of Molecular Sciences 24, no. 2: 1208. https://doi.org/10.3390/ijms24021208
APA StyleCzajlik, A., Batta, Á., Kerner, K., Fizil, Á., Hajdu, D., Raics, M., Kövér, K. E., & Batta, G. (2023). DMSO-Induced Unfolding of the Antifungal Disulfide Protein PAF and Its Inactive Variant: A Combined NMR and DSC Study. International Journal of Molecular Sciences, 24(2), 1208. https://doi.org/10.3390/ijms24021208