

“Pulsed Hypoxia” Gradually Reprograms Breast Cancer Fibroblasts into Pro-Tumorigenic Cells via Mesenchymal–Epithelial Transition

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
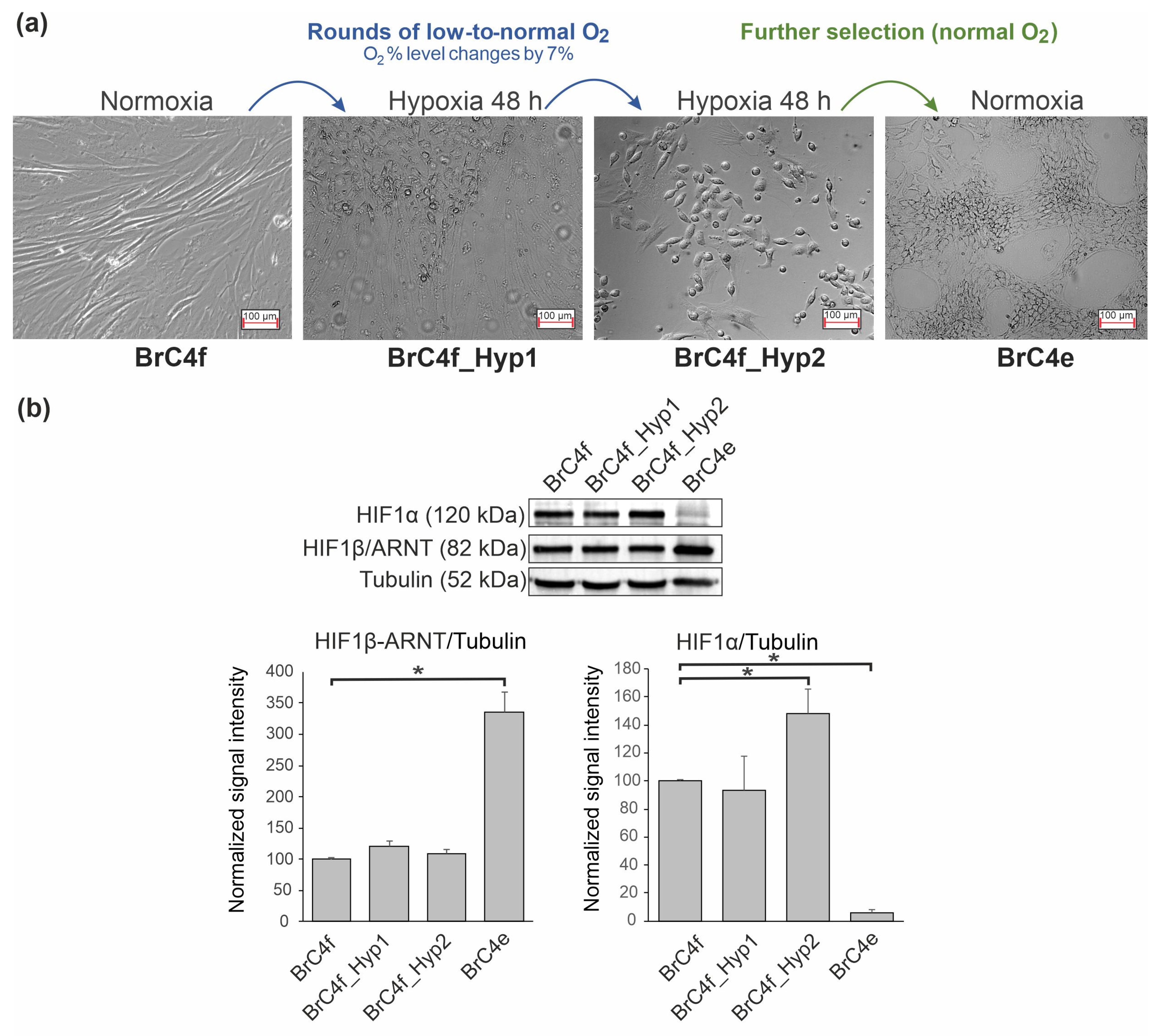
2.1. Hypoxia-Induced Changes in Cell Morphology of a Fibroblast Culture
2.2. PH-Induced Transformation of Fibroblast Follows a Change in Mesenchymal Markers Including Cytoskeletal Markers and an Increase in E-Cadherin
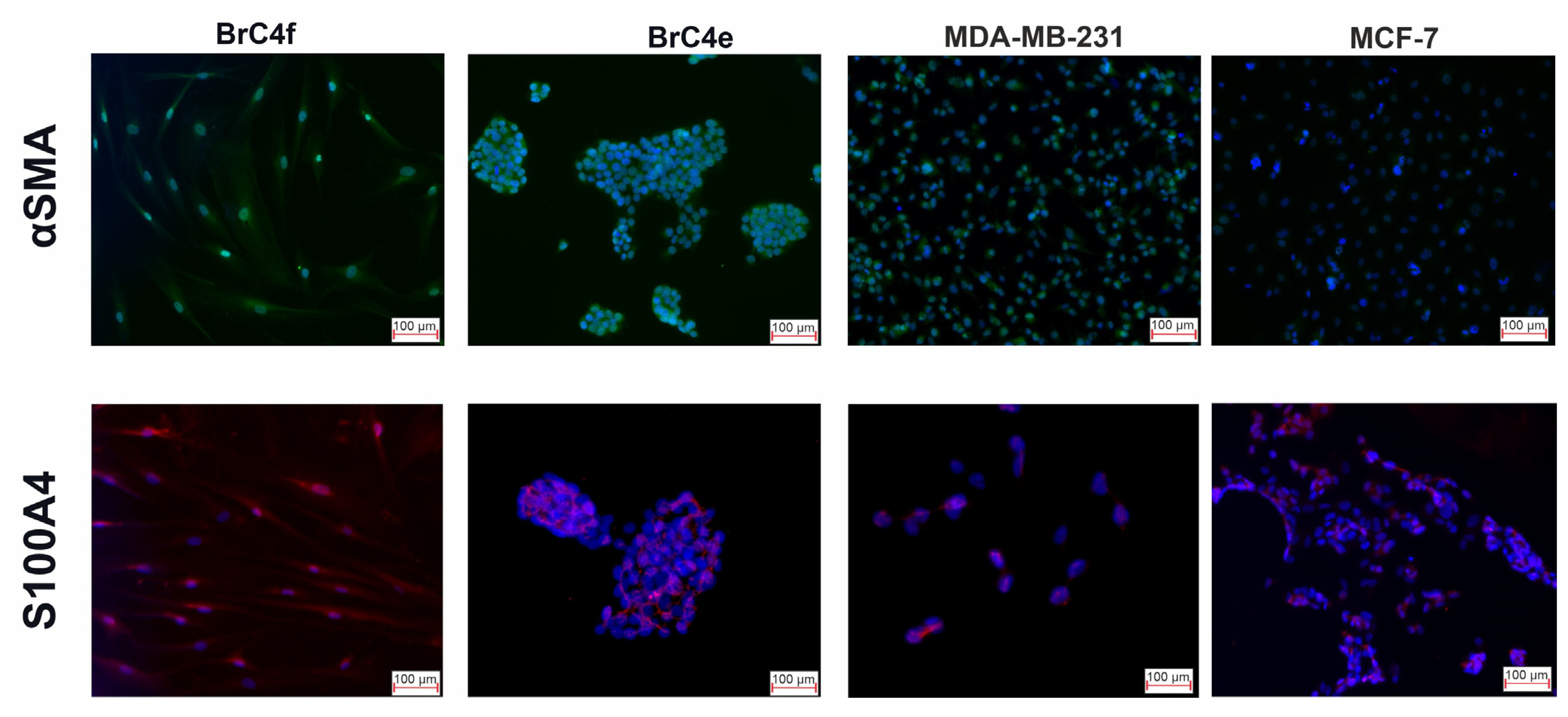
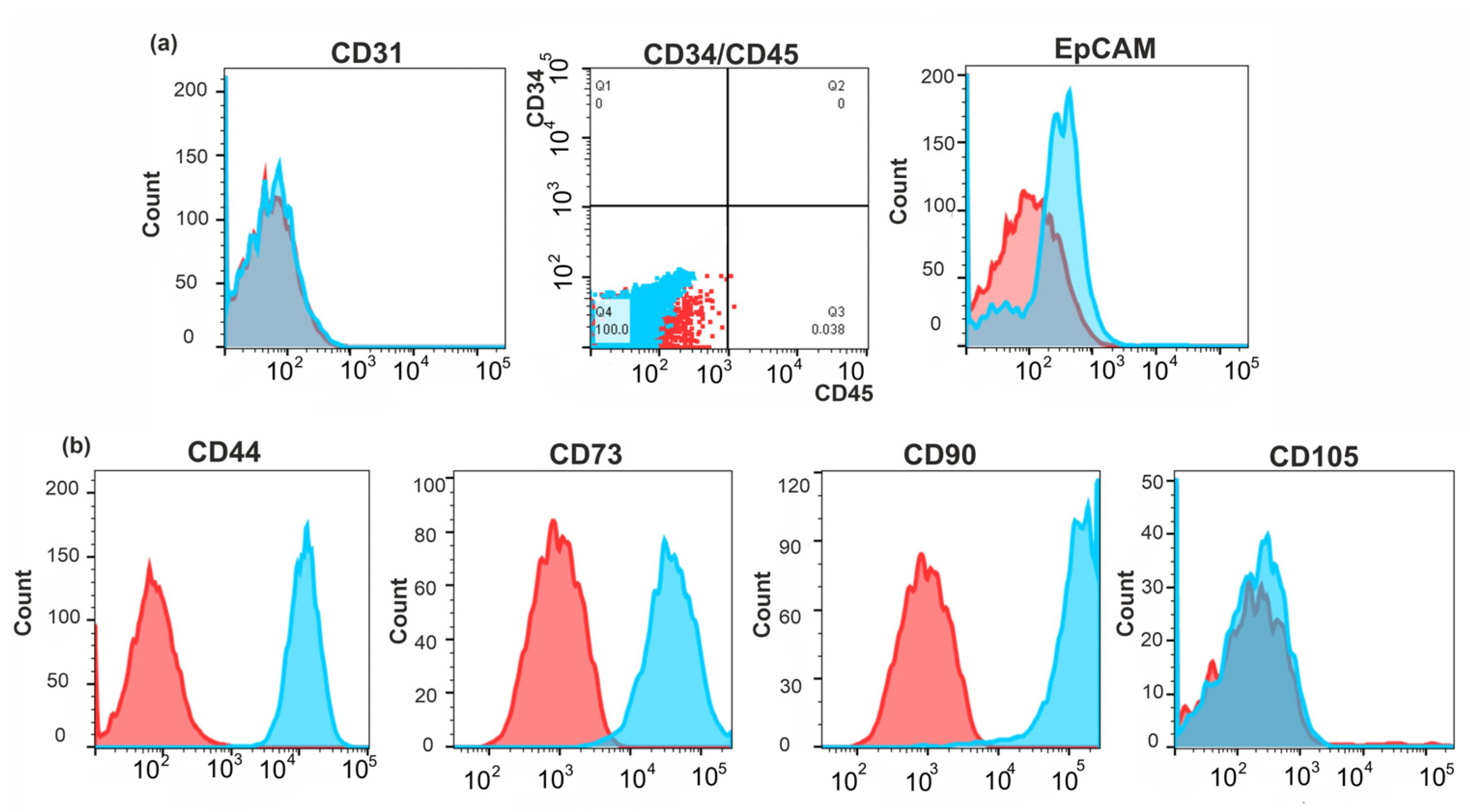
2.3. Identification of the Molecular Markers of Cancer-Associated Fibroblasts in BrC4f Cells
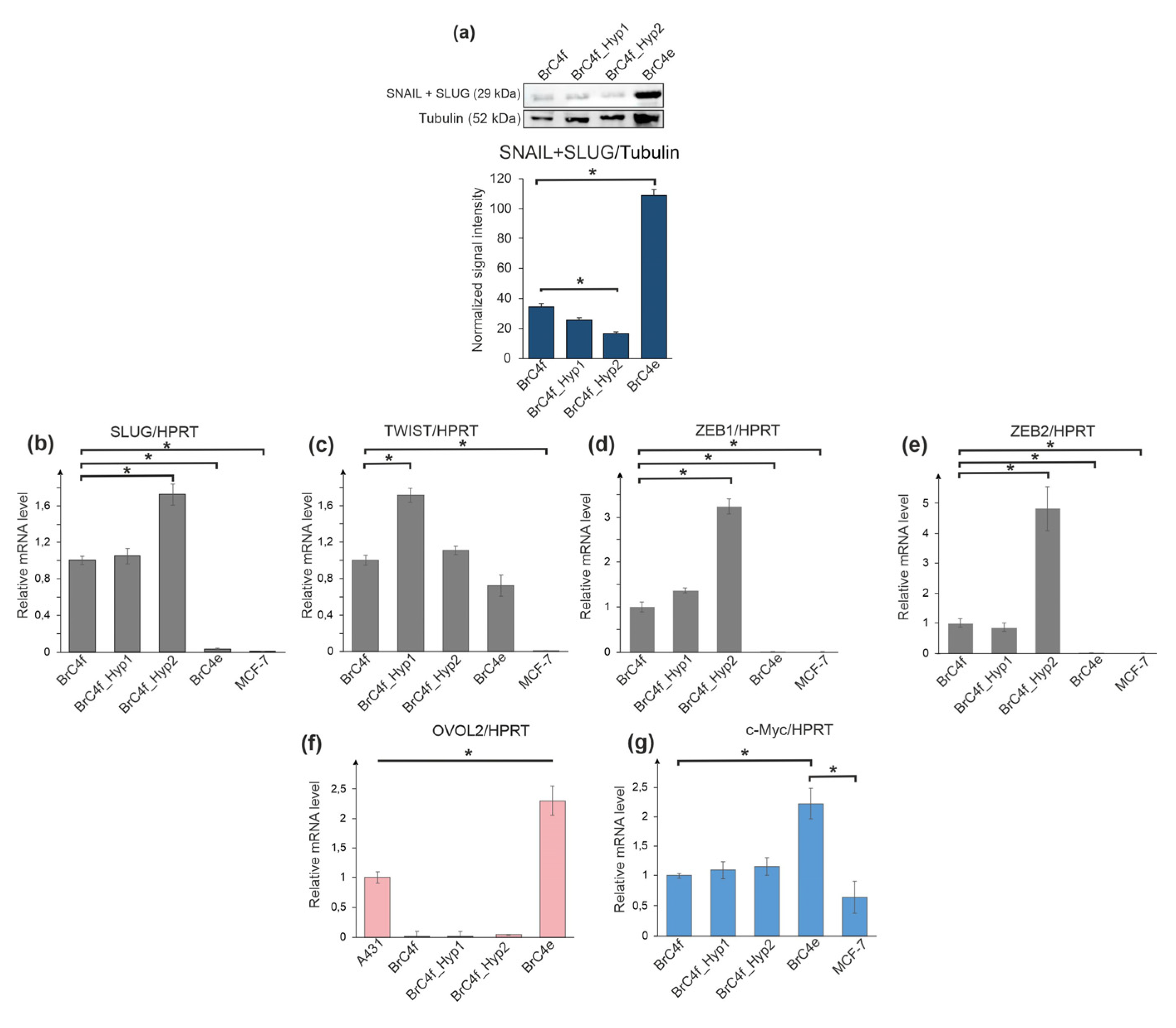
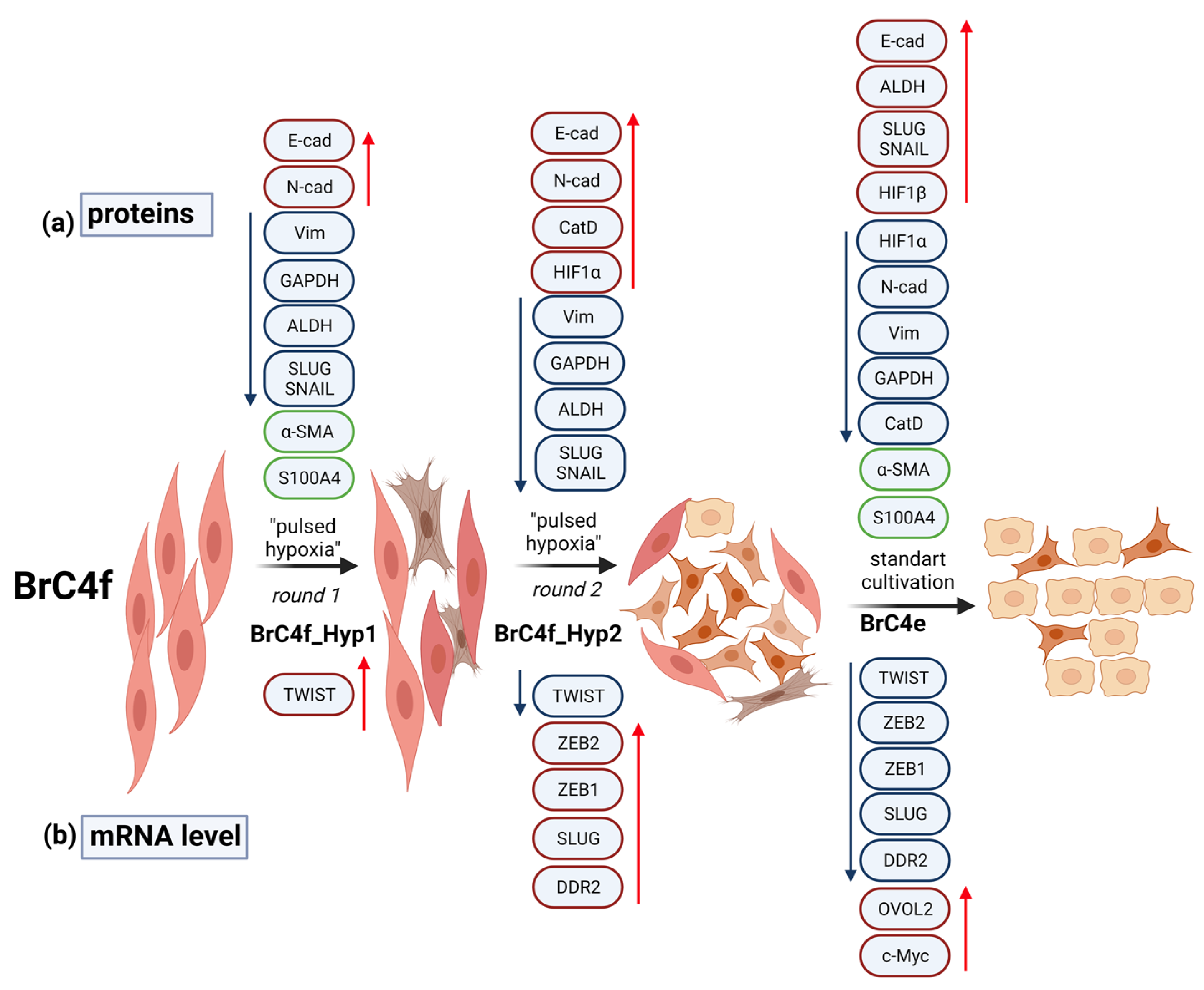
2.4. Alteration of Gene Expression Profile during Epithelial–Mesenchymal Transition and Mesenchymal-Epithelial Transition
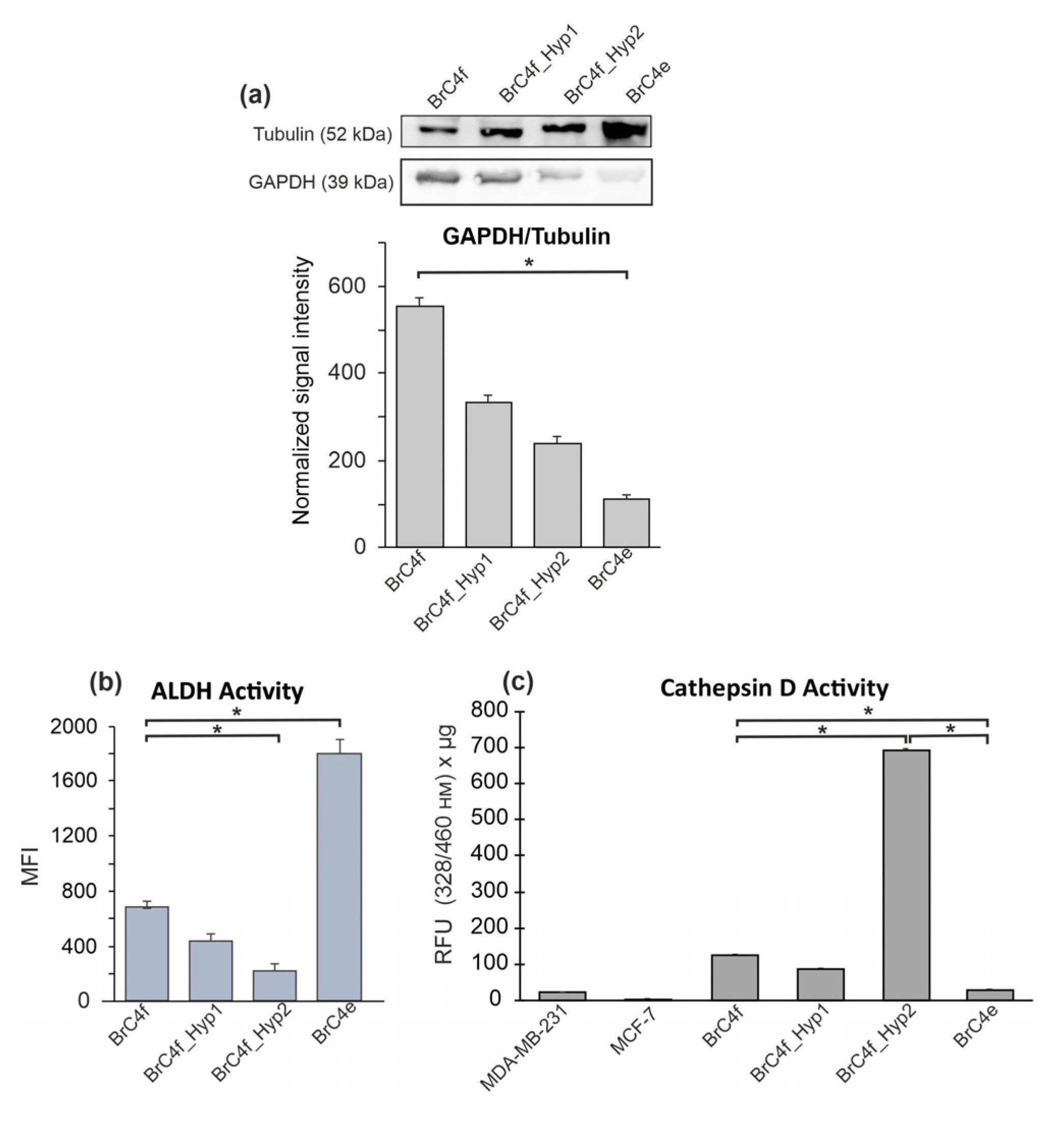
2.5. Mesenchymal–Epithelial Transition Results in the Modifications of Cellular Metabolism
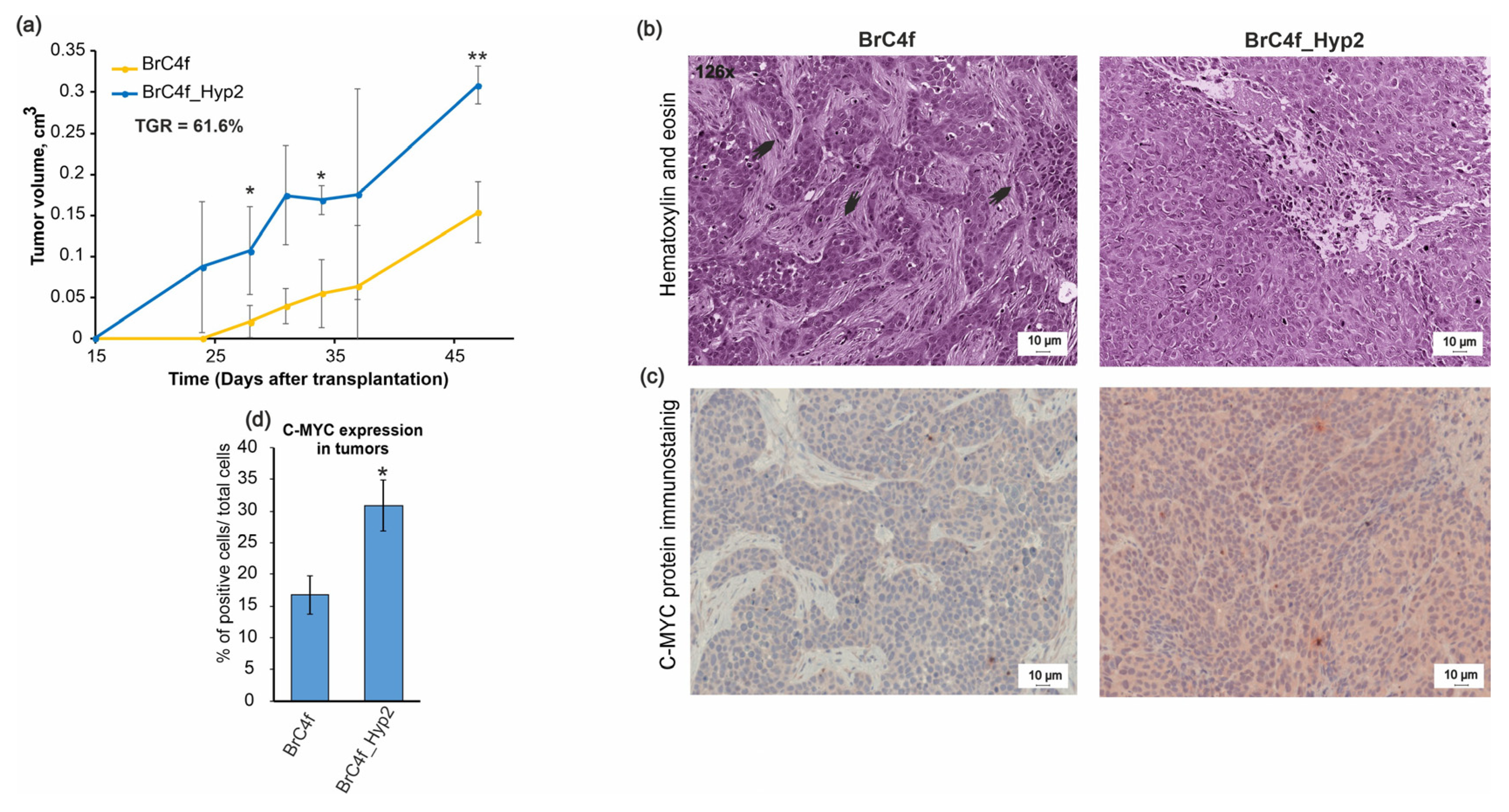
2.6. In Vivo Tumorigenic Properties of Established Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Total RNA Isolation and Reverse Transcriptase–Quantitative Polymerase Chain Reaction (RT-qPCR)
4.3. Western Blot
4.4. Flow Cytometery
4.5. Aldefluor Assay
4.6. Cathepsin D Activity
4.7. Immunocytochemistry
4.8. Xenograft Assay
4.9. Hematoxylin and Eosin Staining
4.10. Tissue Samples Immunostaining
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Riggio, A.I.; Varley, K.E.; Welm, A.L. The lingering mysteries of metastatic recurrence in breast cancer. Br. J. Cancer 2020, 124, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Valyi-Nagy, I.; Heldin, C.H.; Herlyn, M.; Westermark, B. Platelet-derived growth factor (PDGF) in oncogenesis: Development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc. Natl. Acad. Sci. USA 1993, 90, 393–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Ermakov, M.S.; Nushtaeva, A.A.; Richter, V.A.; Koval, O.A. Cancer-associated fibroblasts and their role in tumor progression. Vestn. VOGiS 2022, 26, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Berdiel-Acer, M.; Sanz-Pamplona, R.; Calon, A.; Cuadras, D.; Berenguer, A.; Sanjuan, X.; Paules, M.J.; Salazar, R.; Moreno, V.; Batlle, E.; et al. Differences between CAFs and their paired NCF from adjacent colonic mucosa reveal functional heterogeneity of CAFs, providing prognostic information. Mol. Oncol. 2014, 8, 1290–1305. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, T.; Shen, J.; Li, S.; Lin, J.; Zheng, X.; Gao, Q.; Zhou, H. Separation, cultivation and biological characteristics of oral carcinoma-associated fibroblasts. Oral Dis. 2006, 12, 375–380. [Google Scholar] [CrossRef]
- Mercier, I.; Casimiro, M.C.; Wang, C.; Rosenberg, A.L.; Quong, J.; Minkeu, A.; Allen, K.G.; Danilo, C.; Sotgia, F.; Bonuccelli, G.; et al. Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 down-regulation and RB tumor suppressor functional inactivation: Implications for the response to hormonal therapy. Cancer Biol. Ther. 2008, 7, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Zawieracz, K.; Eckert, M.A. Isolation of Normal and Cancer-Associated Fibroblasts. In Ovarian Cancer; Kreeger, P.K., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2022; Volume 2424, pp. 155–165. ISBN 978-1-07-161955-1. [Google Scholar]
- Han, C.; Liu, T.; Yin, R. Biomarkers for cancer-associated fibroblasts. Biomark. Res. 2020, 8, 64. [Google Scholar] [CrossRef]
- Glabman, R.A.; Choyke, P.L.; Sato, N. Cancer-Associated Fibroblasts: Tumorigenicity and Targeting for Cancer Therapy. Cancers 2022, 14, 3906. [Google Scholar] [CrossRef] [PubMed]
- Elwakeel, E.; Weigert, A. Breast Cancer CAFs: Spectrum of Phenotypes and Promising Targeting Avenues. Int. J. Mol. Sci. 2021, 22, 11636. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Nogueira, P.; Fuster, G.; Gutierrez-Uzquiza, Á.; Gascón, P.; Carbó, N.; Bragado, P. Cancer-Associated Fibroblasts in Breast Cancer Treatment Response and Metastasis. Cancers 2021, 13, 3146. [Google Scholar] [CrossRef]
- Lai, X.; Li, Q.; Wu, F.; Lin, J.; Chen, J.; Zheng, H.; Guo, L. Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons from Somatic Cell Reprogramming. Front. Cell Dev. Biol. 2020, 8, 760. [Google Scholar] [CrossRef]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the Mesenchymal–Epithelial Transition and Its Relationship with Metastatic Tumor Formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef] [Green Version]
- Pei, D.; Shu, X.; Gassama-Diagne, A.; Thiery, J.P. Mesenchymal–epithelial transition in development and reprogramming. Nat. Cell Biol. 2019, 21, 44–53. [Google Scholar] [CrossRef]
- Tsai, J.H.; Yang, J. Epithelial–mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.P.; Marie-Egyptienne, D.T.; Hedley, D.W. Cancer Stem Cells, Hypoxia and Metastasis. Semin. Radiat. Oncol. 2009, 19, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Becerril, C.; Montaño, M.; Cisneros, J.; Mendoza-Milla, C.; Pardo, A.; Ortiz-Quintero, B.; Selman, M.; Ramos, C. Mesenchymal–Epithelial Transition in Fibroblasts of Human Normal Lungs and Interstitial Lung Diseases. Biomolecules 2021, 11, 378. [Google Scholar] [CrossRef]
- Watanabe, M.; Buth, J.E.; Vishlaghi, N.; de la Torre-Ubieta, L.; Taxidis, J.; Khakh, B.S.; Coppola, G.; Pearson, C.A.; Yamauchi, K.; Gong, D.; et al. Self-Organized Cerebral Organoids with Human-Specific Features Predict Effective Drugs to Combat Zika Virus Infection. Cell Rep. 2017, 21, 517–532. [Google Scholar] [CrossRef]
- Alotaibi, H.; Basilicata, M.F.; Shehwana, H.; Kosowan, T.; Schreck, I.; Braeutigam, C.; Konu, O.; Brabletz, T.; Stemmler, M.P. Enhancer cooperativity as a novel mechanism underlying the transcriptional regulation of E-cadherin during mesenchymal to epithelial transition. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2015, 1849, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Romero, Y.; Aquino-Gálvez, A. Hypoxia in Cancer and Fibrosis: Part of the Problem and Part of the Solution. Int. J. Mol. Sci. 2021, 22, 8335. [Google Scholar] [CrossRef]
- Henke, N.; Ferreirós, N.; Geisslinger, G.; Ding, M.G.; Essler, S.; Fuhrmann, D.C.; Geis, T.; Namgaladze, D.; Dehne, N.; Brüne, B. Loss of HIF-1β in macrophages attenuates AhR/ARNT-mediated tumorigenesis in a PAH-driven tumor model. Oncotarget 2016, 7, 25915–25929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Semenza, G.L. Purification and Characterization of Hypoxia-inducible Factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A Mesenchymal-to-Epithelial Transition Initiates and Is Required for the Nuclear Reprogramming of Mouse Fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitsidianaki, I.; Morgan, J.; Adams, J.; Campbell, K. Mesenchymal-to-epithelial transitions require tissue-specific interactions with distinct laminins. J. Cell Biol. 2021, 220, e202010154. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Roca, H.; Hernandez, J.; Weidner, S.; McEachin, R.C.; Fuller, D.; Sud, S.; Schumann, T.; Wilkinson, J.E.; Zaslavsky, A.; Li, H.; et al. Transcription Factors OVOL1 and OVOL2 Induce the Mesenchymal to Epithelial Transition in Human Cancer. PLoS ONE 2013, 8, e76773. [Google Scholar] [CrossRef]
- Qi, X.-K.; Han, H.-Q.; Zhang, H.-J.; Xu, M.; Li, L.; Chen, L.; Xiang, T.; Feng, Q.-S.; Kang, T.; Qian, C.-N.; et al. OVOL2 links stemness and metastasis via fine-tuning epithelial-mesenchymal transition in nasopharyngeal carcinoma. Theranostics 2018, 8, 2202–2216. [Google Scholar] [CrossRef]
- Ito, T.; Tsuji, G.; Ohno, F.; Nakahara, T.; Uchi, H.; Furue, M. Potential role of the OVOL1–OVOL2 axis and c-Myc in the progression of cutaneous squamous cell carcinoma. Mod. Pathol. 2017, 30, 919–927. [Google Scholar] [CrossRef] [Green Version]
- Hugo, H.J.; Kokkinos, M.I.; Blick, T.; Ackland, M.L.; Thompson, E.W.; Newgreen, D.F. Defining the E-Cadherin Repressor Interactome in Epithelial-Mesenchymal Transition: The PMC42 Model as a Case Study. Cells Tissues Organs 2011, 193, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Monkman, J.; Blick, T.; Duijf, P.H.; Nagaraj, S.H.; Thompson, E.W. Multi-Omics Characterization of the Spontaneous Mesenchymal–Epithelial Transition in the PMC42 Breast Cancer Cell Lines. J. Clin. Med. 2019, 8, 1253. [Google Scholar] [CrossRef] [Green Version]
- Nushtaeva, A.A.; Karpushina, A.A.; Ermakov, M.S.; Gulyaeva, L.F.; Gerasimov, A.V.; Sidorov, S.V.; Gayner, T.A.; Yunusova, A.Y.; Tkachenko, A.V.; Richter, V.A.; et al. Establishment of primary human breast cancer cell lines using “pulsed hypoxia” method and development of metastatic tumor model in immunodeficient mice. Cancer Cell Int. 2019, 19, 46. [Google Scholar] [CrossRef]
- Schulze, A.B.; Schmidt, L.H.; Heitkötter, B.; Huss, S.; Mohr, M.; Marra, A.; Hillejan, L.; Görlich, D.; Barth, P.J.; Rehkämper, J.; et al. Prognostic impact of CD34 and SMA in cancer-associated fibroblasts in stage I–III NSCLC. Thorac. Cancer 2020, 11, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.-S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J.; et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 515. [Google Scholar] [CrossRef] [Green Version]
- Huynh, P.T.; Beswick, E.J.; Coronado, Y.A.; Johnson, P.; O’Connell, M.R.; Watts, T.; Singh, P.; Qiu, S.; Morris, K.; Powell, D.W.; et al. CD90+ stromal cells are the major source of IL-6, which supports cancer stem-like cells and inflammation in colorectal cancer: Fibroblast Derived IL-6 Supports CRC. Int. J. Cancer 2016, 138, 1971–1981. [Google Scholar] [CrossRef] [Green Version]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021, 39, 1227–1244. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; He, Z.; Yi, B.; Xue, L.; Sun, J. Simvastatin Suppresses Human Breast Cancer Cell Invasion by Decreasing the Expression of Pituitary Tumor-Transforming Gene 1. Front. Pharmacol. 2020, 11, 574068. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, R.; Fujita, K.; Takahashi, S.; Yoneda, H.; Nagao, K.; Masuda, W.; Naito, M.; Tsuruo, T.; Miyatake, K.; Inui, H.; et al. Hypoxia up-regulates glyceraldehyde-3-phosphate dehydrogenase in mouse brain capillary endothelial cells: Involvement of Na+/Ca2+ exchanger. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2003, 1593, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Zou, B.; Peng, S.; Li, P.; Zhu, G.; Chen, J.; Chen, J.; Liu, X.; Zhou, W.; Ding, L.; et al. Nuclear GAPDH is vital for hypoxia-induced hepatic stellate cell apoptosis and is indicative of aggressive hepatocellular carcinoma behavior. Cancer Manag. Res. 2019, 11, 4947–4956. [Google Scholar] [CrossRef] [Green Version]
- Flahaut, M.; Jauquier, N.; Chevalier, N.; Nardou, K.; Balmas Bourloud, K.; Joseph, J.-M.; Barras, D.; Widmann, C.; Gross, N.; Renella, R.; et al. Aldehyde dehydrogenase activity plays a Key role in the aggressive phenotype of neuroblastoma. BMC Cancer 2016, 16, 781. [Google Scholar] [CrossRef] [Green Version]
- Nejad, A.E.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Javanmard, S.H.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Gunasinghe, N.P.A.D.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal–epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Ziani, L.; Buart, S.; Chouaib, S.; Thiery, J. Hypoxia increases melanoma-associated fibroblasts immunosuppressive potential and inhibitory effect on T cell-mediated cytotoxicity. Oncoimmunology 2021, 10, 1950953. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, C.-H.; Tan, L.-D.; Wang, Q.-S.; Li, X.-Q.; Feng, Y.-M. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, S.B.; Dewhirst, M.W.; Hammond, E.M. Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer. Cancers 2020, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Montagna, C. Chromosomal rearrangements in cancer: Detection and Potential Causal Mechanisms. Mol. Cell. Oncol. 2014, 1, e29904. [Google Scholar] [CrossRef] [Green Version]
- Hollestelle, A.; Peeters, J.K.; Smid, M.; Timmermans, M.; Verhoog, L.C.; Westenend, P.; Heine, A.A.J.; Chan, A.; Sieuwerts, A.M.; Wiemer, E.; et al. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 2013, 138, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Song, P.; Li, D.; Yi, C.; Chen, H.; Ruan, S.; Shi, Z.; Xu, W.; Fu, X.; Zheng, S. Cancer-associated fibroblasts from invasive breast cancer have an attenuated capacity to secrete collagens. Int. J. Oncol. 2014, 45, 1479–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corver, W.E.; ter Haar, N.T.; Fleuren, G.J.; Oosting, J. Cervical carcinoma-associated fibroblasts are DNA diploid and do not show evidence for somatic genetic alterations. Cell. Oncol. 2011, 34, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.-H.; Liu, Y.; Zhou, H.-M.; Chen, Q.-M.; Li, B.-Q. Analysis of chromosome karyotype of oral carcinoma-associated fibroblasts. Hua Xi Kou Qiang Yi Xue Za Zhi 2005, 23, 159–160. [Google Scholar] [PubMed]
- Baudoin, N.; Bloomfield, M. Karyotype Aberrations in Action: The Evolution of Cancer Genomes and the Tumor Microenvironment. Genes 2021, 12, 558. [Google Scholar] [CrossRef]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Attieh, Y.; Barbazan, J.; et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grum-Schwensen, B.; Klingelhofer, J.; Berg, C.H.; El-Naaman, C.; Grigorian, M.; Lukanidin, E.; Ambartsumian, N. Suppression of Tumor Development and Metastasis Formation in Mice Lacking the S100A4(mts1) Gene. Cancer Res 2005, 65, 3772–3780. [Google Scholar] [CrossRef] [Green Version]
- Corsa, C.A.; Brenot, A.; Grither, W.R.; Van Hove, S.; Loza, A.J.; Zhang, K.; Ponik, S.M.; Liu, Y.; DeNardo, D.G.; Eliceiri, K.W.; et al. The Action of Discoidin Domain Receptor 2 in Basal Tumor Cells and Stromal Cancer-Associated Fibroblasts Is Critical for Breast Cancer Metastasis. Cell Rep. 2016, 15, 2510–2523. [Google Scholar] [CrossRef] [Green Version]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef] [PubMed]
- Mandl, M.; Depping, R. Hypoxia-Inducible Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT) (HIF-1β): Is It a Rare Exception? Mol. Med. 2014, 20, 215–220. [Google Scholar] [CrossRef]
- Zhang, Y.; Bian, Y.; Wang, Y.; Wang, Y.; Duan, X.; Han, Y.; Zhang, L.; Wang, F.; Gu, Z.; Qin, Z. HIF-1α is necessary for activation and tumour-promotion effect of cancer-associated fibroblasts in lung cancer. J. Cell. Mol. Med. 2021, 25, 5457–5469. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701. [Google Scholar] [CrossRef]
- Kim, J.-W.; Evans, C.; Weidemann, A.; Takeda, N.; Lee, Y.S.; Stockmann, C.; Branco-Price, C.; Brandberg, F.; Leone, G.; Ostrowski, M.C.; et al. Loss of Fibroblast HIF-1α Accelerates Tumorigenesis. Cancer Res 2012, 72, 3187–3195. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [Green Version]
- Lundgren, K.; Nordenskjöld, B.; Landberg, G. Hypoxia, Snail and incomplete epithelial–mesenchymal transition in breast cancer. Br. J. Cancer 2009, 101, 1769–1781. [Google Scholar] [CrossRef]
- Garcia, D.D.; Yarden, Y. Abstract 991: OVO-like genes are master regulators of the epithelial fate in breast cancer. Cancer Res 2022, 82, 991. [Google Scholar] [CrossRef]
- Wu, J.; Luo, D.; Li, S. Ovo Like Zinc Finger 2 (OVOL2) Suppresses Breast Cancer Stem Cell Traits and Correlates with Immune Cells Infiltration. Breast Cancer: Targets Ther. 2022, 14, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashimura, Y.; Nakajima, Y.; Yamaji, R.; Harada, N.; Shibasaki, F.; Nakano, Y.; Inui, H. Up-regulation of glyceraldehyde-3-phosphate dehydrogenase gene expression by HIF-1 activity depending on Sp1 in hypoxic breast cancer cells. Arch. Biochem. Biophys. 2011, 509, 1–8. [Google Scholar] [CrossRef]
- Vasiliou, V.; Pappa, A.; Estey, T. Role of Human Aldehyde Dehydrogenases in Endobiotic and Xenobiotic Metabolism. Drug Metab. Rev. 2004, 36, 279–299. [Google Scholar] [CrossRef]
- Shiraishi, A.; Tachi, K.; Essid, N.; Tsuboi, I.; Nagano, M.; Kato, T.; Yamashita, T.; Bando, H.; Hara, H.; Ohneda, O. Hypoxia promotes the phenotypic change of aldehyde dehydrogenase activity of breast cancer stem cells. Cancer Sci. 2017, 108, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Ketterer, S.; Mitschke, J.; Ketscher, A.; Schlimpert, M.; Reichardt, W.; Baeuerle, N.; Hess, M.E.; Metzger, P.; Boerries, M.; Peters, C.; et al. Cathepsin D deficiency in mammary epithelium transiently stalls breast cancer by interference with mTORC1 signaling. Nat. Commun. 2020, 11, 5133. [Google Scholar] [CrossRef]
- Balachander, G.M.; Talukdar, P.M.; Debnath, M.; Rangarajan, A.; Chatterjee, K. Inflammatory Role of Cancer-Associated Fibroblasts in Invasive Breast Tumors Revealed Using a Fibrous Polymer Scaffold. ACS Appl. Mater. Interfaces 2018, 10, 33814–33826. [Google Scholar] [CrossRef]
- Li, Y.; Wei, Y.; Tang, W.; Luo, J.; Wang, M.; Lin, H.; Guo, H.; Ma, Y.; Zhang, J.; Li, Q. Association between the degree of fibrosis in fibrotic focus and the unfavorable clinicopathological prognostic features of breast cancer. PeerJ 2019, 7, e8067. [Google Scholar] [CrossRef] [Green Version]
- Mayrhofer, R.M.; Ng, H.P.; Putti, T.C.; Kuchel, P.W. Magnetic Resonance in the Detection of Breast Cancers of Different Histological Types. Magn. Reson. Insights 2013, 6, MRI.S10640. [Google Scholar] [CrossRef]
- Shi, J.-W.; Liu, W.; Zhang, T.-T.; Wang, S.; Lin, X.-L.; Li, J.; Jia, J.-S.; Sheng, H.-F.; Yao, Z.-F.; Zhao, W.-T.; et al. The enforced expression of c-Myc in pig fibroblasts triggers mesenchymal-epithelial transition (MET) via F-actin reorganization and RhoA/Rock pathway inactivation. Cell Cycle 2013, 12, 1119–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef]
- Qiu, X.; Boufaied, N.; Hallal, T.; Feit, A.; de Polo, A.; Luoma, A.M.; Alahmadi, W.; Larocque, J.; Zadra, G.; Xie, Y.; et al. MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets. Nat. Commun. 2022, 13, 2559. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Parvani, J.G.; Schiemann, W.P. The Pathophysiology of Epithelial-Mesenchymal Transition Induced by Transforming Growth Factor-β in Normal and Malignant Mammary Epithelial Cells. J. Mammary Gland. Biol. Neoplasia 2010, 15, 169–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Schmidt, B.U.S.; Kubitschke, H.; Morawetz, E.W.; Wolf, B.; Käs, J.A.; Losert, W. Detecting heterogeneity in and between breast cancer cell lines. Cancer Converg. 2020, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Y.; Zhou, K.; Zhang, G.; Wang, F.; Ren, J. Isolation and Characterization of CD105+/CD90+ Subpopulation in Breast Cancer MDA-MB-231 Cell Line. Int. J. Clin. Exp. Pathol. 2015, 8, 5105–5112. [Google Scholar] [PubMed]
- Boon, K. The Mesenchymal-Like Phenotype of the MDA-MB-231 Cell Line. In Breast Cancer—Focusing Tumor Microenvironment, Stem cells and Metastasis; Gunduz, M., Ed.; InTechOpen: London, UK, 2011; ISBN 978-953-307-766-6. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Sequence 5‘→3‘ | Primer Length |
|---|---|---|
| TWIST1 | F: GGCATCACTATGGACTTTCTCTATT R: GGCCAGTTTGATCCCAGTATT | 25 21 |
| SLUG | F: TGGTTGCTTCAAGGACACAT R: GCAAATGCTCTGTTGCAGTG | 20 20 |
| ZEB2 | F: CGATCCAGACCGCAATTAAC R: TGCTGACTGCATGACCATC | 20 19 |
| ZEB1 | F: AACTGCTGGGAGGATGACAC R: TCCTGCTTCATCTGCCTGA | 20 19 |
| OVOL1 | F: ACGATGCCCATCCACTACCTG R: TTTCTGAGGTGCTGGTCATCATTC | 21 24 |
| OVOL2 | F: GGCAAGGGCTTCAACGACA R: CTTCAGGTGGGACTCCAGAGA | 19 21 |
| C-MYC | F: CTTCTCTCCGTCCTCGGATTCT R: GAAGGTGATCCAGACTCTGACCTT | 22 24 |
| HPRT | F: CATCAAAGCACTGAATAGAAAT R: TATCTTCCACAATCAAGACATT | 22 22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nushtaeva, A.; Ermakov, M.; Abdurakhmanova, M.; Troitskaya, O.; Belovezhets, T.; Varlamov, M.; Gayner, T.; Richter, V.; Koval, O. “Pulsed Hypoxia” Gradually Reprograms Breast Cancer Fibroblasts into Pro-Tumorigenic Cells via Mesenchymal–Epithelial Transition. Int. J. Mol. Sci. 2023, 24, 2494. https://doi.org/10.3390/ijms24032494
Nushtaeva A, Ermakov M, Abdurakhmanova M, Troitskaya O, Belovezhets T, Varlamov M, Gayner T, Richter V, Koval O. “Pulsed Hypoxia” Gradually Reprograms Breast Cancer Fibroblasts into Pro-Tumorigenic Cells via Mesenchymal–Epithelial Transition. International Journal of Molecular Sciences. 2023; 24(3):2494. https://doi.org/10.3390/ijms24032494
Chicago/Turabian StyleNushtaeva, Anna, Mikhail Ermakov, Maria Abdurakhmanova, Olga Troitskaya, Tatyana Belovezhets, Mikhail Varlamov, Tatyana Gayner, Vladimir Richter, and Olga Koval. 2023. "“Pulsed Hypoxia” Gradually Reprograms Breast Cancer Fibroblasts into Pro-Tumorigenic Cells via Mesenchymal–Epithelial Transition" International Journal of Molecular Sciences 24, no. 3: 2494. https://doi.org/10.3390/ijms24032494
APA StyleNushtaeva, A., Ermakov, M., Abdurakhmanova, M., Troitskaya, O., Belovezhets, T., Varlamov, M., Gayner, T., Richter, V., & Koval, O. (2023). “Pulsed Hypoxia” Gradually Reprograms Breast Cancer Fibroblasts into Pro-Tumorigenic Cells via Mesenchymal–Epithelial Transition. International Journal of Molecular Sciences, 24(3), 2494. https://doi.org/10.3390/ijms24032494