

Biallelic Variants in TULP1 Are Associated with Heterogeneous Phenotypes of Retinal Dystrophy
 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Clinical findings
2.1.1. Leber Congenital Amaurosis (LCA)
2.1.2. Early-Onset Retinitis Pigmentosa (eoRP)
2.1.3. Retinitis Pigmentosa (RP)
2.1.4. Cone Dystrophy (CD)
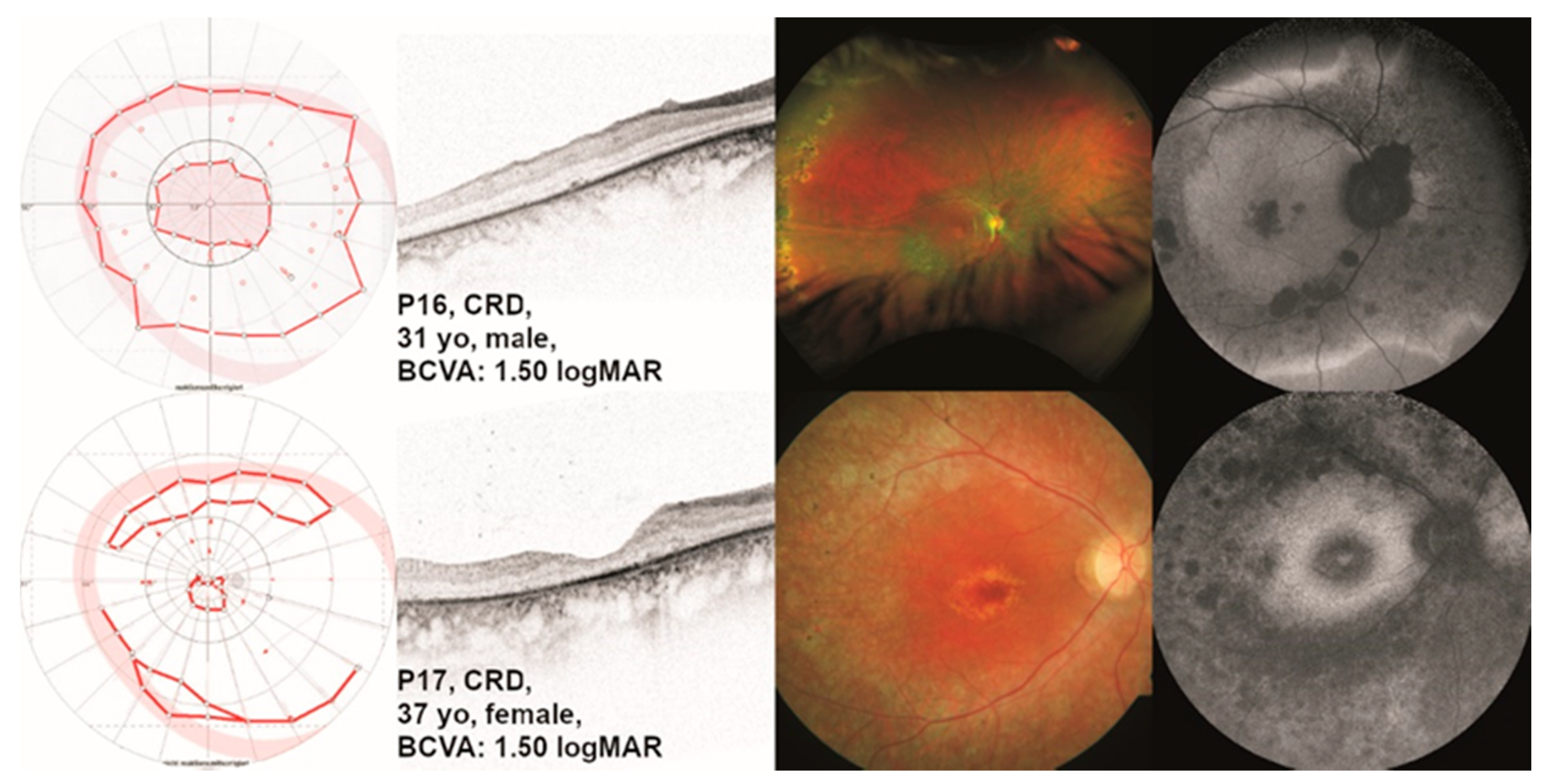
2.1.5. Cone-Rod Dystrophy (CRD)
2.2. Genetic Findings and Minigene Assays
2.3. Molecular Modeling of Structural Variants
3. Discussion
4. Materials and Methods
4.1. Ophthalmological Testing
4.2. Genetic Diagnostic Testing
4.3. In Vitro Splice Assays
4.4. Molecular Modeling of Human TULP1 and Structural Analysis of Pathological Variants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Puech, B.; Kostrubiec, B.; Hache, J.C.; François, P. Epidemiology and prevalence of hereditary retinal dystrophies in the Northern France. J. Fr. Ophtalmol. 1991, 14, 153–164. [Google Scholar] [PubMed]
- Congdon, N.; O’Colmain, B.; Klaver, C.C.; Klein, R.; Muñoz, B.; Friedman, D.S.; Kempen, J.; Taylor, H.R.; Mitchell, P.; Eye Diseases Prevalence Research Group. Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 2004, 122, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef]
- Holtan, J.P.; Selmer, K.K.; Heimdal, K.R.; Bragadóttir, R. Inherited retinal disease in Norway—A characterization of current clinical and genetic knowledge. Acta Ophthalmol. 2020, 98, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, S.A.; North, M.A.; Nishina, P.L.; Berson, E.L.; Dryja, T.P. Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with retinitis pigmentosa. Nat. Genet. 1998, 18, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Kabir, F.; Iqbal, M.; Gottsch, C.B.; Naeem, M.A.; Assir, M.Z.; Khan, S.N.; Akram, J.; Riazuddin, S.; Ayyagari, R.; et al. Pathogenic mutations in TULP1 responsible for retinitis pigmentosa identified in consanguineous familial cases. Mol. Vis. 2016, 22, 797–815. [Google Scholar]
- den Hollander, A.I.; van Lith-Verhoeven, J.J.; Arends, M.L.; Strom, T.M.; Cremers, F.P.; Hoyng, C.B. Novel compound heterozygous TULP1 mutations in a family with severe early-onset retinitis pigmentosa. Arch. Ophthalmol. 2007, 125, 932–935. [Google Scholar] [CrossRef] [Green Version]
- Souzeau, E.; Thompson, J.A.; McLaren, T.L.; De Roach, J.N.; Barnett, C.P.; Lamey, T.M.; Craig, J.E. Maternal uniparental isodisomy of chromosome 6 unmasks a novel variant in TULP1 in a patient with early onset retinal dystrophy. Mol. Vis. 2018, 24, 478–484. [Google Scholar]
- Mataftsi, A.; Schorderet, D.F.; Chachoua, L.; Boussalah, M.; Nouri, M.T.; Barthelmes, D.; Borruat, F.X.; Munier, F.L. Novel TULP1 mutation causing leber congenital amaurosis or early onset retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5160–5167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.A.; De Roach, J.N.; McLaren, T.L.; Montgomery, H.E.; Hoffmann, L.H.; Campbell, I.R.; Chen, F.K.; Mackey, D.A.; Lamey, T.M. The genetic profile of Leber congenital amaurosis in an Australian cohort. Mol. Genet. Genom. Med. 2017, 5, 652–667. [Google Scholar] [CrossRef]
- Roosing, S.; van den Born, L.I.; Hoyng, C.B.; Thiadens, A.A.; de Baere, E.; Collin, R.W.; Koenekoop, R.K.; Leroy, B.P.; van Moll-Ramirez, N.; Venselaar, H.; et al. Maternal uniparental isodisomy of chromosome 6 reveals a TULP1 mutation as a novel cause of cone dysfunction. Ophthalmology 2013, 120, 1239–1246. [Google Scholar] [CrossRef]
- Al-Hindi, H.; Chauhan, M.Z.; Sanders, R.; Samarah, H.; DeBenedictis, M.; Traboulsi, E.; Uwaydat, S.H. TULP1 related retinal dystrophy: Report of rare and novel variants with a previously undescribed phenotype in two cases. Ophthalmic Genet. 2022, 43, 277–281. [Google Scholar] [CrossRef]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 2020, 41, 1514–1527. [Google Scholar] [CrossRef]
- Jaffal, L.; Joumaa, H.; Mrad, Z.; Zeitz, C.; Audo, I.; El Shamieh, S. The genetics of rod-cone dystrophy in Arab countries: A systematic review. Eur. J. Hum. Genet. 2021, 29, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, M.A.; Naggert, J.K.; Yan, Y.; Noben-Trauth, K.; Nishina, P.M. Molecular characterization of TUB, TULP1, and TULP2, members of the novel tubby gene family and their possible relation to ocular diseases. Proc. Natl. Acad. Sci. USA 1997, 94, 3128–3133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, S.; He, W.; Ikeda, A.; Naggert, J.K.; North, M.A.; Nishina, P.M. Cell-specific expression of tubby gene family members (tub, Tulp1,2, and 3) in the retina. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2706–2712. [Google Scholar]
- Xi, Q.; Pauer, G.J.; Marmorstein, A.D.; Crabb, J.W.; Hagstrom, S.A. Tubby-like protein 1 (TULP1) interacts with F-actin in photoreceptor cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4754–4761. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Jackson, P.K. The tubby family proteins. Genome Biol. 2011, 12, 225. [Google Scholar] [CrossRef] [Green Version]
- Santagata, S.; Boggon, T.J.; Baird, C.L.; Gomez, C.A.; Zhao, J.; Shan, W.S.; Myszka, D.G.; Shapiro, L. G-protein signaling through tubby proteins. Science 2001, 292, 2041–2050. [Google Scholar] [CrossRef]
- Hagstrom, S.A.; Adamian, M.; Scimeca, M.; Pawlyk, B.S.; Yue, G.; Li, T. A role for the Tubby-like protein 1 in rhodopsin transport. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1955–1962. [Google Scholar]
- Remez, L.; Cohen, B.; Nevet, M.J.; Rizel, L.; Ben-Yosef, T. TULP1 and TUB Are Required for Specific Localization of PRCD to Photoreceptor Outer Segments. Int. J. Mol. Sci. 2020, 21, 8677. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, S.A.; Duyao, M.; North, M.A.; Li, T. Retinal degeneration in tulp1−/− mice: Vesicular accumulation in the interphotoreceptor matrix. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2795–2802. [Google Scholar]
- Hong, J.J.; Kim, K.E.; Park, S.Y.; Bok, J.; Seo, J.T.; Moon, S.J. Differential Roles of Tubby Family Proteins in Ciliary Formation and Trafficking. Mol. Cells 2021, 44, 591–601. [Google Scholar] [CrossRef]
- Xi, Q.; Pauer, G.J.; Ball, S.L.; Rayborn, M.; Hollyfield, J.G.; Peachey, N.S.; Crabb, J.W.; Hagstrom, S.A. Interaction between the photoreceptor-specific tubby-like protein 1 and the neuronal-specific GTPase dynamin-1. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2837–2844. [Google Scholar] [CrossRef] [PubMed]
- Grossman, G.H.; Beight, C.D.; Ebke, L.A.; Pauer, G.J.; Hagstrom, S.A. Interaction of tubby-like protein-1 (Tulp1) and microtubule-associated protein (MAP) 1A and MAP1B in the mouse retina. Adv. Exp. Med. Biol. 2014, 801, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Ebke, L.A.; Sinha, S.; Pauer, G.J.T.; Hagstrom, S.A. Photoreceptor Compartment-Specific TULP1 Interactomes. Int. J. Mol. Sci. 2021, 22, 8066. [Google Scholar] [CrossRef]
- Wahl, S.; Magupalli, V.G.; Dembla, M.; Katiyar, R.; Schwarz, K.; Köblitz, L.; Alpadi, K.; Krause, E.; Rettig, J.; Sung, C.H.; et al. The Disease Protein Tulp1 Is Essential for Periactive Zone Endocytosis in Photoreceptor Ribbon Synapses. J. Neurosci. 2016, 36, 2473–2493. [Google Scholar] [CrossRef] [Green Version]
- Caberoy, N.B. Synergistic interaction of tubby and tubby-like protein 1 (Tulp1). Adv. Exp. Med. Biol. 2014, 801, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boggon, T.J.; Shan, W.S.; Santagata, S.; Myers, S.C.; Shapiro, L. Implication of tubby proteins as transcription factors by structure-based functional analysis. Science 1999, 286, 2119–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palfi, A.; Yesmambetov, A.; Millington-Ward, S.; Shortall, C.; Humphries, P.; Kenna, P.F.; Chadderton, N.; Farrar, G.J. AAV-Delivered Tulp1 Supplementation Therapy Targeting Photoreceptors Provides Minimal Benefit in Tulp1−/− Retinas. Front. Neurosci. 2020, 14, 891. [Google Scholar] [CrossRef]
- Blindness and Vision Impairment. Available online: https://www.who.int/news-room/fact-sheets/detail/blindness-and-visual-impairment (accessed on 10 July 2022).
- Blindheit im Sinne des Gesetzes. Available online: https://www.dbsv.org/iii-der-schwerbehindertenausweis.html (accessed on 10 July 2022).
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Burset, M.; Seledtsov, I.A.; Solovyev, V.V. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000, 28, 4364–4375. [Google Scholar] [CrossRef]
- Gu, S.; Lennon, A.; Li, Y.; Lorenz, B.; Fossarello, M.; North, M.; Gal, A.; Wright, A. Tubby-like protein-1 mutations in autosomal recessive retinitis pigmentosa. Lancet 1998, 351, 1103–1104. [Google Scholar] [CrossRef] [PubMed]
- Comander, J.; Weigel-DiFranco, C.; Maher, M.; Place, E.; Wan, A.; Harper, S.; Sandberg, M.A.; Navarro-Gomez, D.; Pierce, E.A. The Genetic Basis of Pericentral Retinitis Pigmentosa-A Form of Mild Retinitis Pigmentosa. Genes 2017, 8, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampaglione, E.; Kinde, B.; Place, E.M.; Navarro-Gomez, D.; Maher, M.; Jamshidi, F.; Nassiri, S.; Mazzone, J.A.; Finn, C.; Schlegel, D.; et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet. Med. 2020, 22, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, L.; Maltese, P.E.; Castori, M.; El Shamieh, S.; Zeitz, C.; Audo, I.; Zulian, A.; Marinelli, C.; Benedetti, S.; Costantini, A.; et al. Molecular Epidemiology in 591 Italian Probands with Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef] [PubMed]
- Lobo, G.P.; Au, A.; Kiser, P.D.; Hagstrom, S.A. Involvement of Endoplasmic Reticulum Stress in TULP1 Induced Retinal Degeneration. PLoS ONE 2016, 11, e0151806. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, H.; Peng, J.; Gibbs, R.A.; Lewis, R.A.; Lupski, J.R.; Mardon, G.; Chen, R. Mutation survey of known LCA genes and loci in the Saudi Arabian population. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1336–1343. [Google Scholar] [CrossRef]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Hebrard, M.; Manes, G.; Bocquet, B.; Meunier, I.; Coustes-Chazalette, D.; Herald, E.; Senechal, A.; Bolland-Auge, A.; Zelenika, D.; Hamel, C.P. Combining gene mapping and phenotype assessment for fast mutation finding in non-consanguineous autosomal recessive retinitis pigmentosa families. Eur. J. Hum. Genet. 2011, 19, 1256–1263. [Google Scholar] [CrossRef]
- Kannabiran, C.; Singh, H.; Sahini, N.; Jalali, S.; Mohan, G. Mutations in TULP1, NR2E3, and MFRP genes in Indian families with autosomal recessive retinitis pigmentosa. Mol. Vis. 2012, 18, 1165–1174. [Google Scholar] [PubMed]
- Ajmal, M.; Khan, M.I.; Micheal, S.; Ahmed, W.; Shah, A.; Venselaar, H.; Bokhari, H.; Azam, A.; Waheed, N.K.; Collin, R.W.; et al. Identification of recurrent and novel mutations in TULP1 in Pakistani families with early-onset retinitis pigmentosa. Mol. Vis. 2012, 18, 1226–1237. [Google Scholar]
- Lewis, C.A.; Batlle, I.R.; Batlle, K.G.; Banerjee, P.; Cideciyan, A.V.; Huang, J.; Alemán, T.S.; Huang, Y.; Ott, J.; Gilliam, T.C.; et al. Tubby-like protein 1 homozygous splice-site mutation causes early-onset severe retinal degeneration. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2106–2114. [Google Scholar]
- Den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Huang, W.C.; Sumaroka, A.; Roman, A.J.; Schwartz, S.B.; Luo, X.; Sheplock, R.; Dauber, J.M.; Swider, M.; et al. TULP1 mutations causing early-onset retinal degeneration: Preserved but insensitive macular cones. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5354–5364. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.O.; Bergmann, C.; Eisenberger, T.; Bolz, H.J. A TULP1 founder mutation, p.Gln301*, underlies a recognisable congenital rod-cone dystrophy phenotype on the Arabian Peninsula. Br. J. Ophthalmol. 2015, 99, 488–492. [Google Scholar] [CrossRef]
- Chen, X.; Sheng, X.; Liu, Y.; Li, Z.; Sun, X.; Jiang, C.; Qi, R.; Yuan, S.; Wang, X.; Zhou, G.; et al. Distinct mutations with different inheritance mode caused similar retinal dystrophies in one family: A demonstration of the importance of genetic annotations in complicated pedigrees. J. Transl. Med. 2018, 16, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avela, K.; Salonen-Kajander, R.; Laitinen, A.; Ramsden, S.; Barton, S.; Rudanko, S.L. The genetic aetiology of retinal degeneration in children in Finland—New founder mutations identified. Acta Ophthalmol. 2019, 97, 805–814. [Google Scholar] [CrossRef]
- Verbakel, S.K.; Fadaie, Z.; Klevering, B.J.; van Genderen, M.M.; Feenstra, I.; Cremers, F.P.M.; Hoyng, C.B.; Roosing, S. The identification of a RNA splice variant in TULP1 in two siblings with early-onset photoreceptor dystrophy. Mol. Genet. Genom. Med. 2019, 7, e660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodard, D.R.; Xing, C.; Ganne, P.; Liang, H.; Mahindrakar, A.; Sankurathri, C.; Hulleman, J.D.; Mootha, V.V. A novel homozygous missense mutation p.P388S in TULP1 causes protein instability and retinitis pigmentosa. Mol. Vis. 2021, 27, 179–190. [Google Scholar]
- Majander, A.; Sankila, E.M.; Falck, A.; Vasara, L.K.; Seitsonen, S.; Kulmala, M.; Haavisto, A.K.; Avela, K.; Turunen, J.A. Natural history and biomarkers of retinal dystrophy caused by the biallelic TULP1 variant c.148delG. Acta Ophthalmol. 2022; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Diñeiro, M.; Capín, R.; Cifuentes, G.Á.; Fernández-Vega, B.; Villota, E.; Otero, A.; Santiago, A.; Pruneda, P.C.; Castillo, D.; Viejo-Díaz, M.; et al. Comprehensive genomic diagnosis of inherited retinal and optical nerve disorders reveals hidden syndromes and personalized therapeutic options. Acta Ophthalmol. 2020, 98, e1034–e1048. [Google Scholar] [CrossRef]
- Pinelli, M.; Carissimo, A.; Cutillo, L.; Lai, C.H.; Mutarelli, M.; Moretti, M.N.; Singh, M.V.; Karali, M.; Carrella, D.; Pizzo, M.; et al. An atlas of gene expression and gene co-regulation in the human retina. Nucleic Acids Res. 2016, 44, 5773–5784. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Melamud, E.; Moult, J. Stochastic noise in splicing machinery. Nucleic Acids Res. 2009, 37, 4873–4886. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Visual Acuity “Cheat Sheet”—High and Low Vision. Available online: https://michaelbach.de/sci/acuity.html (accessed on 9 June 2022).
- Weisschuh, N.; Mazzola, P.; Heinrich, T.; Haack, T.; Wissinger, B.; Tonagel, F.; Kelbsch, C. First submicroscopic inversion of the OPA1 gene identified in dominant optic atrophy—A case report. BMC Med. Genet. 2020, 21, 236. [Google Scholar] [CrossRef] [PubMed]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisschuh, N.; Wissinger, B.; Gramer, E. A splice site mutation in the PAX6 gene which induces exon skipping causes autosomal dominant inherited aniridia. Mol. Vis. 2012, 18, 751–757. [Google Scholar] [PubMed]
- Weisschuh, N.; Marino, V.; Schäferhoff, K.; Richter, P.; Park, J.; Haack, T.B.; Dell’Orco, D. Mutations at a split codon in the GTPase-encoding domain of OPA1 cause dominant optic atrophy through different molecular mechanisms. Hum. Mol. Genet. 2022, 31, 761–774. [Google Scholar] [CrossRef]
- Scotto-Lavino, E.; Du, G.; Frohman, M.A. 3’ end cDNA amplification using classic RACE. Nat. Protoc. 2006, 1, 2742–2745. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Clinical Diagnosis | Age at Last Exam | Sex | BCVA [logMAR] | VF Radius [Target III4e] | ERG | TULP1 Genotype |
|---|---|---|---|---|---|---|---|
| P1 | LCA | 6 | f | OD 1.15 OS 0.90 | OD n.d. OS ~40° | flat | c.568G>T;p.(E190*) hom |
| P2 | LCA | 12 | f | OD 0.70 OS 0.50 | OD ~10° OS n.d. | flat | c.901C>T;p.(Q301*) hom |
| P3 | LCA | 62 | f | OD 2.30 OS 2.30 | OD n.d. OS n.d. | flat | c.1523G>A;p.(R508H) hom |
| P4 | eoRP | 6 | m | OD 0.40 OS 0.40 | OD n.d. OS n.d. | n.d. | c.1163C>A;p.(P388Q) het c.1445G>A;p.(R482Q) het |
| P5 | eoRP | 14 | m | OD 0.50 OS 0.40 | OD n.d. OS ~40° | flat | c.629C>G;p.(S210*) hom |
| P6 | eoRP | 21 | m | OD 0.50 OS 0.50 | OD <10° OS <10° | n.d. | c.1081C>T;p.(R361*) het c.1258C>A;p.(R420S) het |
| P7 | eoRP | 25 | f | OD 0.40 OS 0.60 | OD <10° OS <10° | n.d. | c.1081C>T;p.(R361*) het c.1258C>A;p.(R420S) het |
| P8 | eoRP | 33 | m | OD 1.30 OS 1.20 | OD ~10° OS ~10° | n.d. | c.1025G>A;p.(R342Q) het c.1496-6C>A;p.(P499Lfs*143) het |
| P9 | eoRP | 34 | m | OD 2.30 OS 2.30 | OD n.d. OS n.d. | flat | c.1495+1G>A;p.(A442Pfs*18) hom |
| P10 | eoRP | 37 | m | OD 0.60 OS 0.70 | OD <10° OS <10° | flat | c.1047T>G;p.(N349K) hom |
| P11 | eoRP | 37 | m | OD 1.50 OS 0.60 | OD <10° OS < 10° | flat | c.1047T>G;p.(N349K) hom |
| P12 | eoRP | 38 | f | OD 0.80 OS 0.70 | OD n.d. OS n.d. | flat | c.1507_1521del; p.(V503_G507del) hom |
| P13 | RP | 18 | f | OD 0.10 OS 0.10 | OD n.d. OS ~10° | flat | c.1024C>T;p.(R342*) het c.1496-6C>A; p.(P499Lfs*143) het |
| P14 | RP | 80 | m | OD 2.70 OS 2.30 | OD n.d. OS n.d. | flat | c.1496-6C>A;p.(P499Lfs*143) hom |
| P15 | CD | 40 | m | OD 0.40 OS 0.40 | OD normal external boundaries OS normal external boundaries | dark-adapted within normal limits, light-adapted reduced | c.1201C>T;p.(Q401*) het c.797G>T;p.(G266V) het |
| P16 | CRD | 31 | m | OD 1.50 OS 1.30 | OD central scotoma OS central scotoma | dark-adapted slightly reduced, light-adapted reduced | c.1163C>A;p.(P388Q) hom |
| P17 | CRD | 37 | f | OD 1.50 OS 2.30 | OD pericentral scotoma OS pericentral scotoma | flat | c.1471T>C; p.(F491L) het c.1496-6C>A; p.(P499Lfs*143) het |
| cDNA Position (NM_003322.6) | Amino Acid Position (NP_003313.3) | Variant Class | HGMD Accession Number | gnomAD MAF | Observed Number of Alleles in Cohort |
|---|---|---|---|---|---|
| c.568G>T | p.(E190*) | nonsense | - | - | 2 |
| c.629C>G | p.(S210*) | nonsense | CM140491 | - | 2 |
| c.797G>T | p.(G266V) | missense | CM2035471 | 0.0007141 | 1 |
| c.901C>T | p.(Q301*) | nonsense | CM098172 | - | 2 |
| c.1024C>T | p.(R342*) | nonsense | - | - | 1 |
| c.1025G>A | p.(R342Q) | missense | CM119412 | 0.00001988 | 1 |
| c.1047T>G | p.(N349K) | missense | CM123537 | 0.000007953 | 4 |
| c.1081C>T | p.(R361*) | nonsense | CM140477 | 0.000003977 | 2 |
| c.1163C>A | p.(P388Q) | missense | - | - | 3 |
| c.1201C>T | p.(Q401*) | nonsense | CM2034386 | - | 1 |
| c.1258C>A | p.(R420S) | missense | CM135101 | 0.000008988 | 2 |
| c.1445G>A | p.(R482Q) | missense | CM123565 | 0.000003977 | 1 |
| c.1471T>C | p.(F491L) | missense | CM981971 | - | 1 |
| c.1495+1G>A | p.(A442Pfs*18) | splice site | CS982391 | 0.00001195 | 2 |
| c.1496-6C>A/p.? | p.(P499Lfs*143) | splice site | CS984713 | 0.00008210 | 5 |
| c.1507_1521del | p.(V503_G507del) | In-frame deletion | - | - | 2 |
| c.1523G>A | p.(R508H) | missense | CM2036799 | 0.00001066 | 2 |
| Variant [Reference] | Apo | IP3-Bound | |
|---|---|---|---|
| ∆∆Gfapp (kcal/mol) | ∆∆Gfapp (kcal/mol) | IP3 ∆∆Gbapp (kcal/mol) | |
| R342Q[46] | 6.97 ± 3.45 | 4.49 ± 1.53 | 1.10 ± 0.16 |
| N349K [47] | 11.55 ± 0.04 | 40.10 ± 26.59 | −1.24 ± 0.26 |
| P388Q | 9.53 ± 0.30 | 17.97 ± 3.30 | 2.67 ± 3.77 |
| R420S [11] | 15.58 ± 5.81 | 14.79 ± 3.46 | 2.96 ± 2.60 |
| R482Q [48] | 24.32 ± 0.04 | 29.43 ± 1.70 | 2.69 ± 1.99 |
| F491L [5] | 5.44 ± 0.88 | 3.58 ± 1.05 | 0.11 ± 0.03 |
| R508H | 29.32 ± 9.02 | 29.26 ± 8.29 | 13.54 ± 3.30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bodenbender, J.-P.; Marino, V.; Bethge, L.; Stingl, K.; Haack, T.B.; Biskup, S.; Kohl, S.; Kühlewein, L.; Dell’Orco, D.; Weisschuh, N. Biallelic Variants in TULP1 Are Associated with Heterogeneous Phenotypes of Retinal Dystrophy. Int. J. Mol. Sci. 2023, 24, 2709. https://doi.org/10.3390/ijms24032709
Bodenbender J-P, Marino V, Bethge L, Stingl K, Haack TB, Biskup S, Kohl S, Kühlewein L, Dell’Orco D, Weisschuh N. Biallelic Variants in TULP1 Are Associated with Heterogeneous Phenotypes of Retinal Dystrophy. International Journal of Molecular Sciences. 2023; 24(3):2709. https://doi.org/10.3390/ijms24032709
Chicago/Turabian StyleBodenbender, Jan-Philipp, Valerio Marino, Leon Bethge, Katarina Stingl, Tobias B. Haack, Saskia Biskup, Susanne Kohl, Laura Kühlewein, Daniele Dell’Orco, and Nicole Weisschuh. 2023. "Biallelic Variants in TULP1 Are Associated with Heterogeneous Phenotypes of Retinal Dystrophy" International Journal of Molecular Sciences 24, no. 3: 2709. https://doi.org/10.3390/ijms24032709
APA StyleBodenbender, J. -P., Marino, V., Bethge, L., Stingl, K., Haack, T. B., Biskup, S., Kohl, S., Kühlewein, L., Dell’Orco, D., & Weisschuh, N. (2023). Biallelic Variants in TULP1 Are Associated with Heterogeneous Phenotypes of Retinal Dystrophy. International Journal of Molecular Sciences, 24(3), 2709. https://doi.org/10.3390/ijms24032709