
The Autotaxin-LPA Axis Emerges as a Novel Regulator of Smooth Muscle Cell Phenotypic Modulation during Intimal Hyperplasia

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
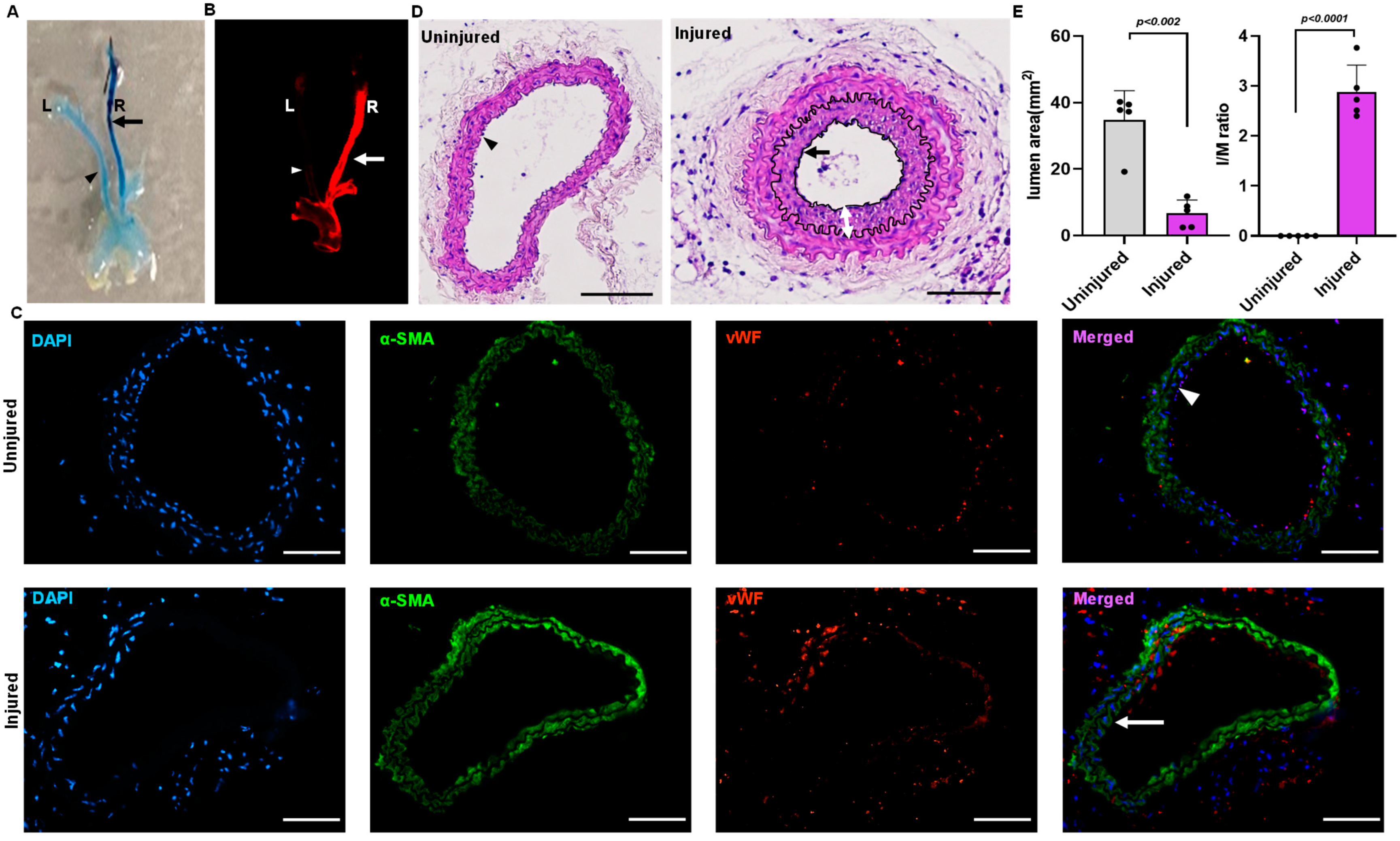
2.1. Mouse Model for Endothelial Denudation
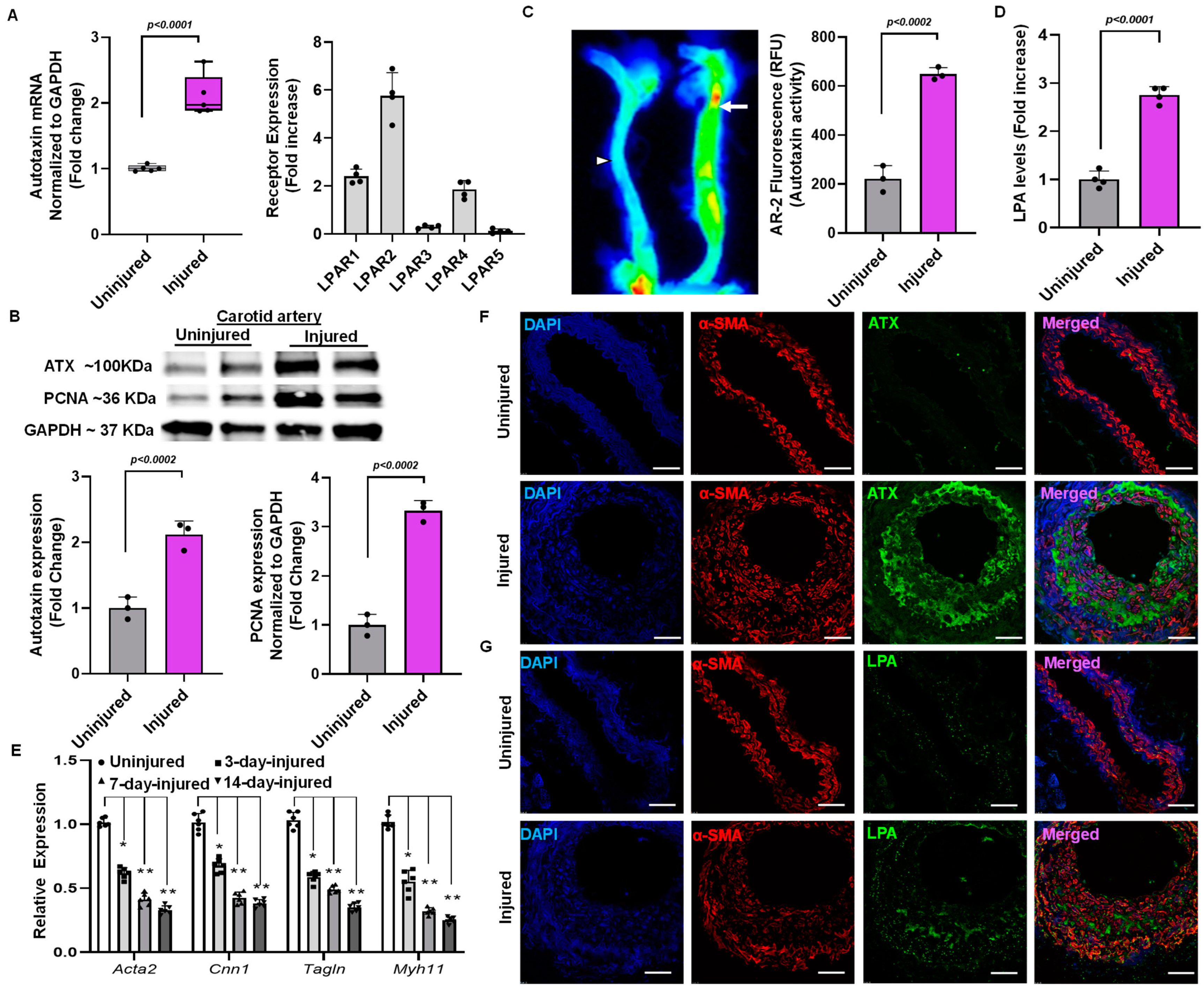
2.2. Elevation of ATX Activity and Expression Following Endothelial Denudation Injury
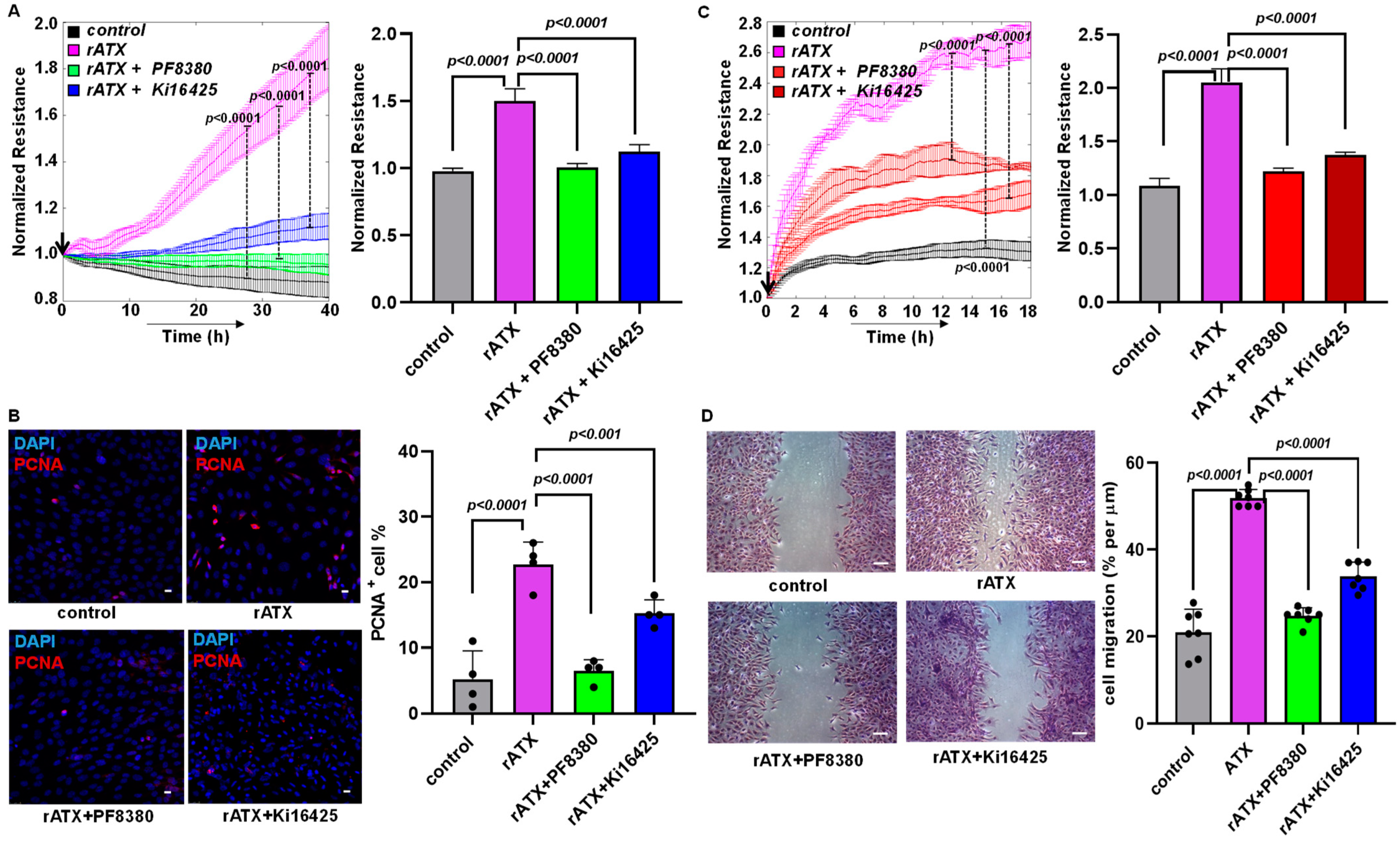
2.3. ATX Inhibition Decreased VSMC Proliferation and Migration
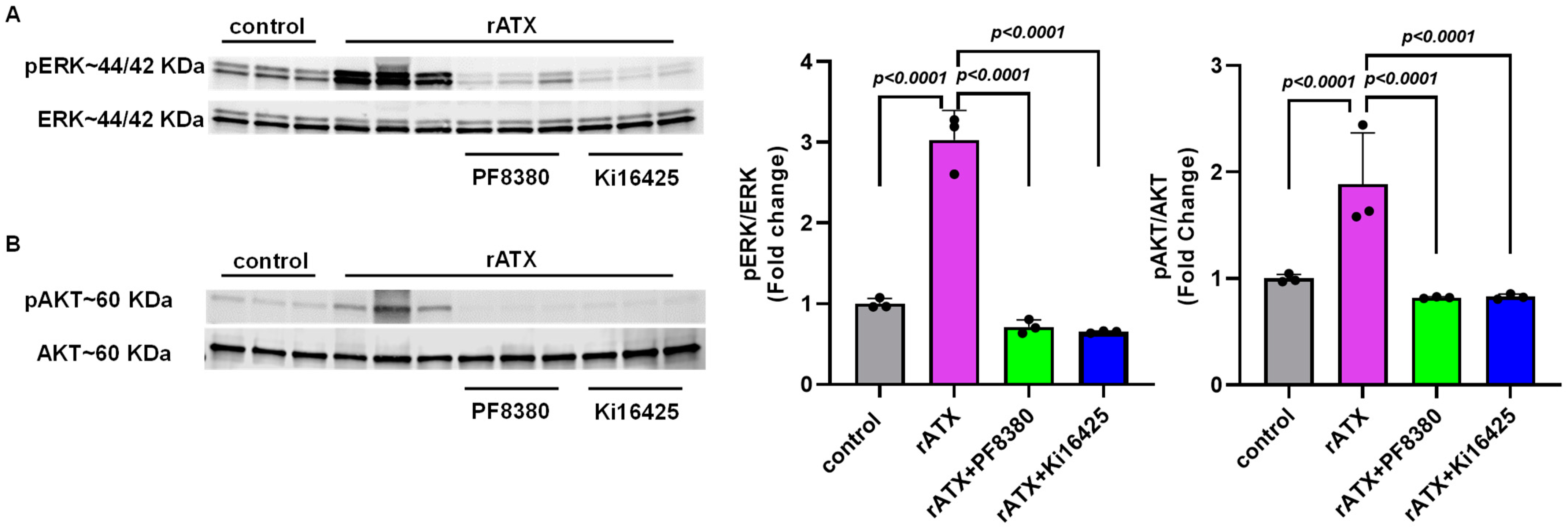
2.4. ATX-Mediated Phosphorylation of AKT–ERK Signaling in VSMCs
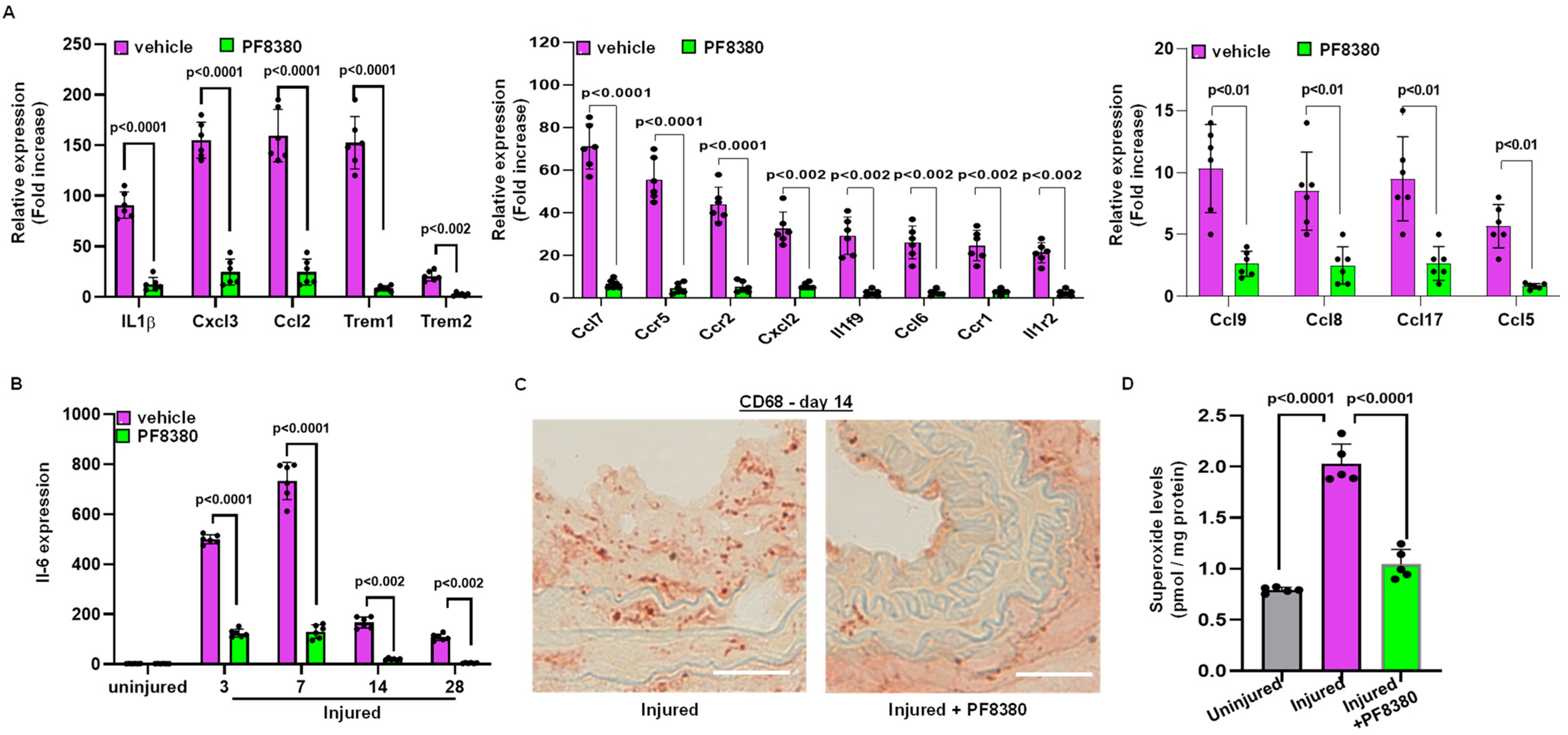
2.5. ATX Inhibitor Reduces Inflammation Following Injury
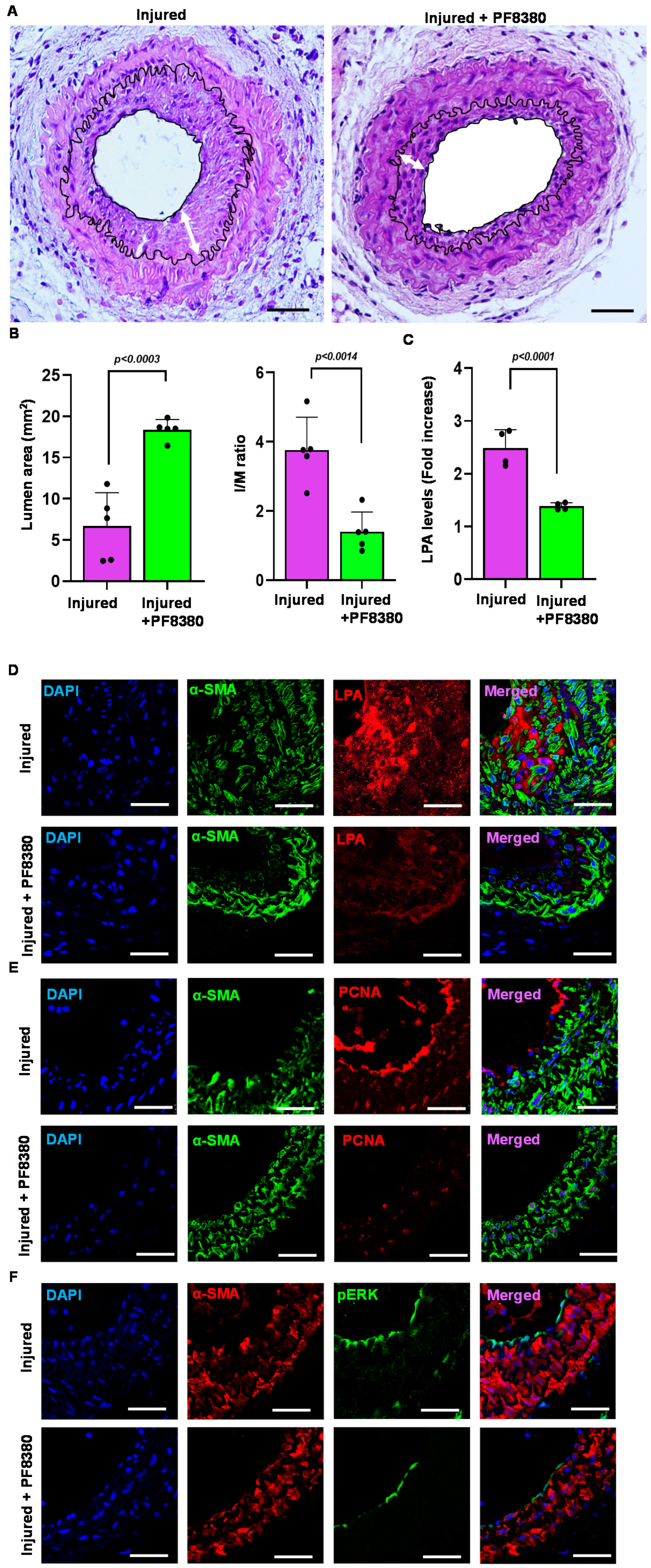
2.6. ATX Inhibitor Reduces Neointimal Hyperplasia
3. Discussion
4. Methods and Materials
4.1. Animal Model of Vascular Injury
4.2. Cell Culture
4.3. ATX Activity Assay
4.4. Migration and Proliferation Assay Using an Electric Cell–Substrate Impedance Sensor (ECIS)
4.5. Hematoxylin and Eosin Staining
4.6. Wound Healing Assay (Scratch Assay)
4.7. Western Blot Analysis
4.8. Immunofluorescence Staining for Cell Proliferation
4.9. Immunohistochemistry
4.10. Real-Time PCR mRNA Expression of LPA Receptors
4.11. Superoxide Measurement
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cui, C.; Huang, X.; Liu, X.; Li, W.; Lu, X.; Lu, M.; Jiang, M.; Yin, M. Endovascular treatment of atherosclerotic popliteal artery disease based on dynamic angiography findings. J. Vasc. Surg. 2017, 65, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Geng, S.; Dai, Y. Therapeutic effect of vascular interventional therapy and aspirin combined with defibrase on cerebral ischemia in rats. Exp. Med. 2018, 16, 891–895. [Google Scholar] [CrossRef]
- Schillinger, M.; Minar, E. Restenosis after percutaneous angioplasty: The role of vascular inflammation. Vasc. Health Risk Manag. 2005, 1, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Achim, A.; Lackó, D.; Hüttl, A.; Csobay-Novák, C.; Csavajda, Á.; Sótonyi, P.; Merkely, B.; Nemes, B.; Ruzsa, Z. Impact of Diabetes Mellitus on Early Clinical Outcome and Stent Restenosis after Carotid Artery Stenting. J. Diabetes Res. 2022, 2022, 4196195. [Google Scholar] [CrossRef]
- Zain, M.A.; Jamil, R.T.; Siddiqui, W.J. Neointimal hyperplasia. In StatPearls [Internet]; StatPearls Publishing: Tampa, FL, USA, 2021. [Google Scholar]
- Rai, V.; Touré, F.; Chitayat, S.; Pei, R.; Song, F.; Li, Q.; Zhang, J.; Rosario, R.; Ramasamy, R.; Chazin, W.J.; et al. Lysophosphatidic acid targets vascular and oncogenic pathways via RAGE signaling. J. Exp. Med. 2012, 209, 2339–2350. [Google Scholar] [CrossRef]
- van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin-LPA axis. Prog. Lipid Res. 2007, 46, 145–160. [Google Scholar] [CrossRef]
- Aoki, J. [Mechanism of lysophosphatidic acid production]. Seikagaku 2003, 75, 609–613. [Google Scholar] [CrossRef]
- Lin, M.-E.; Herr, D.R.; Chun, J. Lysophosphatidic acid (LPA) receptors: Signaling properties and disease relevance. Prostaglandins Other Lipid Mediat. 2010, 91, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.-W.; Mutoh, T.; Lin, M.-E.; Teo, S.T.; Park, K.E.; Mosley, A.N. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef]
- Sugden, M.; Holness, M. Lysophosphatidic acid effects on atherosclerosis and thrombosis. Clin. Lipidol. 2011, 6, 413–426. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Nishida, W.; Hayashi, K.I.; Ohkawa, Y.; Ogawa, A.; Aoki, J.; Arai, H.; Sobue, K. Vascular remodeling induced by naturally occurring unsaturated lysophosphatidic acid in vivo. Circulation 2003, 108, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Yung, Y.C.; Chen, A.; Chun, J. Lysophosphatidic acid signalling in development. Development 2015, 142, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Smyth, S.S.; Cheng, H.-Y.; Miriyala, S.; Panchatcharam, M.; Morris, A.J. Roles of lysophosphatidic acid in cardiovascular physiology and disease. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2008, 1781, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Siess, W.; Tigyi, G. Thrombogenic and atherogenic activities of lysophosphatidic acid. J. Cell. Biochem. 2004, 92, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Rother, E.; Brandl, R.; Baker, D.L.; Goyal, P.; Gebhard, H.; Tigyi, G.; Siess, W. Subtype-selective antagonists of lysophosphatidic acid receptors inhibit platelet activation triggered by the lipid core of atherosclerotic plaques. Circulation 2003, 108, 741–747. [Google Scholar] [CrossRef]
- Siess, W.; Zangl, K.J.; Essler, M.; Bauer, M.; Brandl, R.; Corrinth, C.; Bittman, R.; Tigyi, G.; Aepfelbacher, M. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6931–6936. [Google Scholar] [CrossRef]
- Panchatcharam, M.; Miriyala, S.; Yang, F.; Rojas, M.; End, C.; Vallant, C.; Dong, A.; Lynch, K.; Chun, J.; Morris, A.J. Lysophosphatidic acid receptors 1 and 2 play roles in regulation of vascular injury responses but not blood pressure. Circ. Res. 2008, 103, 662–670. [Google Scholar] [CrossRef]
- Subramanian, P.; Karshovska, E.; Reinhard, P.; Megens, R.T.; Zhou, Z.; Akhtar, S.; Schumann, U.; Li, X.; van Zandvoort, M.; Ludin, C. Lysophosphatidic acid receptors LPA1 and LPA3 promote CXCL12-mediated smooth muscle progenitor cell recruitment in neointima formation. Circ. Res. 2010, 107, 96–105. [Google Scholar] [CrossRef]
- Yang, L.; Kraemer, M.; Fang, X.F.; Angel, P.M.; Drake, R.R.; Morris, A.J.; Smyth, S.S. LPA receptor 4 deficiency attenuates experimental atherosclerosis. J. Lipid Res. 2019, 60, 972–980. [Google Scholar] [CrossRef]
- Tanaka, M.; Okudaira, S.; Kishi, Y.; Ohkawa, R.; Iseki, S.; Ota, M.; Noji, S.; Yatomi, Y.; Aoki, J.; Arai, H. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J. Biol. Chem. 2006, 281, 25822–25830. [Google Scholar] [CrossRef] [Green Version]
- Fotopoulou, S.; Oikonomou, N.; Grigorieva, E.; Nikitopoulou, I.; Paparountas, T.; Thanassopoulou, A.; Zhao, Z.; Xu, Y.; Kontoyiannis, D.L.; Remboutsika, E.; et al. ATX expression and LPA signalling are vital for the development of the nervous system. Dev. Biol. 2010, 339, 451–464. [Google Scholar] [CrossRef]
- Katsifa, A.; Kaffe, E.; Nikolaidou-Katsaridou, N.; Economides, A.N.; Newbigging, S.; McKerlie, C.; Aidinis, V. The Bulk of Autotaxin Activity Is Dispensable for Adult Mouse Life. PLoS ONE 2015, 10, e0143083. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.; Sharma, S.; Subedi, U.; Ara, H.; Shum, A.; Milena, M.; Bhuiyan, M.S.; Kidambi, S.; Sun, H.; Miriyala, S.; et al. The ATX-LPA Axis Regulates Vascular Permeability during Cerebral Ischemic-Reperfusion. Int. J. Mol. Sci. 2022, 23, 4138. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.; Sharma, S.; Ara, H.; Subedi, U.; Sun, G.; Li, C.; Bhuiyan, M.S.; Kevil, C.; Armstrong, W.P.; Minvielle, M.T.; et al. Disrupted Blood-Brain Barrier and Mitochondrial Impairment by Autotaxin-Lysophosphatidic Acid Axis in Postischemic Stroke. J. Am. Heart Assoc. 2021, 10, e021511. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Murph, M.; Panupinthu, N.; Mills, G.B. ATX-LPA receptor axis in inflammation and cancer. Cell Cycle 2009, 8, 3695–3701. [Google Scholar] [CrossRef]
- Chu, X.; Wei, X.; Lu, S.; He, P. Autotaxin-LPA receptor axis in the pathogenesis of lung diseases. Int. J. Clin. Exp. Med. 2015, 8, 17117–17122. [Google Scholar]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef]
- Orosa, B.; Garcia, S.; Conde, C. The autotaxin-lysophosphatidic acid pathway in pathogenesis of rheumatoid arthritis. Eur. J. Pharm. 2015, 765, 228–233. [Google Scholar] [CrossRef]
- Leblanc, R.; Lee, S.-C.; David, M.; Bordet, J.-C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clézardin, P. Interaction of platelet-derived autotaxin with tumor integrin αVβ3 controls metastasis of breast cancer cells to bone. Blood J. Am. Soc. Hematol. 2014, 124, 3141–3150. [Google Scholar] [CrossRef]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Kooi, C.V.; Morris, A.J.; Smyth, S.S. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef]
- Olorundare, O.E.; Peyruchaud, O.; Albrecht, R.M.; Mosher, D.F. Assembly of a fibronectin matrix by adherent platelets stimulated by lysophosphatidic acid and other agonists. Blood J. Am. Soc. Hematol. 2001, 98, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hasse, S.; Zhao, C.; Bourgoin, S.G. Targeting the autotaxin–Lysophosphatidic acid receptor axis in cardiovascular diseases. Biochem. Pharmacol. 2019, 164, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Panchatcharam, M.; Miriyala, S.; Yang, F.; Leitges, M.; Chrzanowska-Wodnicka, M.; Quilliam, L.A.; Anaya, P.; Morris, A.J.; Smyth, S.S. Enhanced proliferation and migration of vascular smooth muscle cells in response to vascular injury under hyperglycemic conditions is controlled by beta3 integrin signaling. Int. J. Biochem. Cell Biol. 2010, 42, 965–974. [Google Scholar] [CrossRef]
- Panchatcharam, M.; Miriyala, S.; Salous, A.; Wheeler, J.; Dong, A.; Mueller, P.; Sunkara, M.; Escalante-Alcalde, D.; Morris, A.J.; Smyth, S.S. Lipid phosphate phosphatase 3 negatively regulates smooth muscle cell phenotypic modulation to limit intimal hyperplasia. Arter. Thromb. Vasc. Biol. 2013, 33, 52–59. [Google Scholar] [CrossRef]
- Gerthoffer, W.T. Mechanisms of vascular smooth muscle cell migration. Circ. Res. 2007, 100, 607–621. [Google Scholar] [CrossRef]
- Bai, H.; Wu, H.; Zhang, L.; Sun, P.; Liu, Y.; Xie, B.; Zhang, C.; Wei, S.; Wang, W.; Li, J. Adventitial injection of HA/SA hydrogel loaded with PLGA rapamycin nanoparticle inhibits neointimal hyperplasia in a rat aortic wire injury model. Drug Deliv. Transl. Res. 2022, 12, 2950–2959. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Liao, Y.; Liu, Z.; Xu, X.; Sun, W.; Qin, H.; Wang, H.; Liu, J.; Jing, T. Exosomes Derived from AT2R-Overexpressing BMSC Prevent Restenosis After Carotid Artery Injury by Attenuating the Injury-Induced Neointimal Hyperplasia. J. Cardiovasc. Transl. Res. 2022; online ahead of print. [Google Scholar]
- Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The role of vascular smooth muscle cells in arterial remodeling: Focus on calcification-related processes. Int. J. Mol. Sci. 2019, 20, 5694. [Google Scholar] [CrossRef]
- Marx, S.O.; Totary-Jain, H.; Marks, A.R. Vascular smooth muscle cell proliferation in restenosis. Circ. Cardiovasc. Interv. 2011, 4, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the endothelial barrier: Identifying and reconciling controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Larifla, L.; Déprez, I.; Pham, I.; Rideau, D.; Louzier, V.; Adam, M.; Eloit, M.; Foucan, L.; Adnot, S.; Teiger, E. Inhibition of vascular smooth muscle cell proliferation and migration in vitro and neointimal hyperplasia in vivo by adenoviral-mediated atrial natriuretic peptide delivery. J. Gene Med. 2012, 14, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.; Karnovsky, M.J.; Edelman, E.R. Inhibition of experimental neointimal hyperplasia and thrombosis depends on the type of vascular injury and the site of drug administration. Circulation 1993, 88, 1215–1221. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Song, H.; Wei, M. Effect of Lp (a) on human mesangial cell proliferation, adhesion and migration. Zhonghua Er Ke Za Zhi Chin. J. Pediatr. 2004, 42, 734–736. [Google Scholar]
- Amaral, R.F.; Geraldo, L.H.; Einicker-Lamas, M.; e Spohr, T.C.L.d.S.; Mendes, F.; Lima, F.R.S. Microglial lysophosphatidic acid promotes glioblastoma proliferation and migration via LPA1 receptor. J. Neurochem. 2021, 156, 499–512. [Google Scholar] [CrossRef]
- Lin, C.-I.; Chen, C.-N.; Huang, M.-T.; Lee, S.-J.; Lin, C.-H.; Chang, C.-C.; Lee, H. Lysophosphatidic acid upregulates vascular endothelial growth factor-C and tube formation in human endothelial cells through LPA1/3, COX-2, and NF-κB activation-and EGFR transactivation-dependent mechanisms. Cell. Signal. 2008, 20, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, W.; Wei, D.; Hu, K.; Wu, X.; Yao, Y. Effect of the LPA-mediated CXCL12-CXCR4 axis in the tumor proliferation, migration and invasion of ovarian cancer cell lines. Oncol. Lett. 2014, 7, 1581–1585. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Keys, J.R.; Eckhart, A.D. Vascular smooth muscle migration and proliferation in response to lysophosphatidic acid (LPA) is mediated by LPA receptors coupling to Gq. Cell. Signal. 2006, 18, 1695–1701. [Google Scholar] [CrossRef]
- Gaaya, A.; Poirier, O.; Mougenot, N.; Hery, T.; Atassi, F.; Marchand, A.; Saulnier-Blache, J.-S.; Amour, J.; Vogt, J.; Lompré, A.-M. Plasticity-related gene-1 inhibits lysophosphatidic acid-induced vascular smooth muscle cell migration and proliferation and prevents neointima formation. Am. J. Physiol.-Cell Physiol. 2012, 303, C1104–C1114. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.-Z.; Laag, E.; Sun, L.; Tan, M.; Zhao, G.; Xu, X. Lysophosphatidic acid induces early growth response gene 1 expression in vascular smooth muscle cells: CRE and SRE mediate the transcription. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Gierse, J.; Thorarensen, A.; Beltey, K.; Bradshaw-Pierce, E.; Cortes-Burgos, L.; Hall, T.; Johnston, A.; Murphy, M.; Nemirovskiy, O.; Ogawa, S. A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J. Pharmacol. Exp. Ther. 2010, 334, 310–317. [Google Scholar] [CrossRef]
- Benesch, M.G.; Tang, X.; Maeda, T.; Ohhata, A.; Zhao, Y.Y.; Kok, B.P.; Dewald, J.; Hitt, M.; Curtis, J.M.; McMullen, T.P. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J. 2014, 28, 2655–2666. [Google Scholar] [CrossRef]
- Boucharaba, A.; Serre, C.-M.; Guglielmi, J.; Bordet, J.-C.; Clézardin, P.; Peyruchaud, O. The type 1 lysophosphatidic acid receptor is a target for therapy in bone metastases. Proc. Natl. Acad. Sci. USA 2006, 103, 9643–9648. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.N.; Jefferson, L.S.; Kimball, S.R. ERK and Akt signaling pathways function through parallel mechanisms to promote mTORC1 signaling. Am. J. Physiol. Cell Physiol. 2011, 300, C1172–C1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-Y.; Kim, K.-H.; Lee, W.-R.; An, H.-J.; Lee, S.-J.; Han, S.-M.; Lee, K.-G.; Park, Y.-Y.; Kim, K.-S.; Lee, Y.-S. Apamin inhibits PDGF-BB-induced vascular smooth muscle cell proliferation and migration through suppressions of activated Akt and Erk signaling pathway. Vasc. Pharmacol. 2015, 70, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Son, D.J.; Ha, S.J.; Song, H.S.; Lim, Y.; Yun, Y.P.; Lee, J.W.; Moon, D.C.; Park, Y.H.; Park, B.S.; Song, M.J. Melittin inhibits vascular smooth muscle cell proliferation through induction of apoptosis via suppression of nuclear factor-κB and Akt activation and enhancement of apoptotic protein expression. J. Pharmacol. Exp. Ther. 2006, 317, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kim, M.J.; Woo, K.-J.; Hong, J.; Kim, S.-H.; Kim, T.-J. Evening Primrose Extracts Inhibit PDGF-BB-Induced Vascular Smooth Muscle Cell Proliferation and Migration by Regulating Cell-Cycle-Related Proteins. Curr. Issues Mol. Biol. 2022, 44, 1928–1940. [Google Scholar] [CrossRef]
- Zhang, F.; Ren, X.; Zhao, M.; Zhou, B.; Han, Y. Angiotensin-(1–7) abrogates angiotensin II-induced proliferation, migration and inflammation in VSMCs through inactivation of ROS-mediated PI3K/Akt and MAPK/ERK signaling pathways. Sci. Rep. 2016, 6, 34621. [Google Scholar] [CrossRef]
- Huang, L.; Qian, W.; Xu, Y.; Guo, Z.; Yin, Y.; Guo, F.; Zhu, W.; Li, Y. Mesenteric adipocyte contributes to intestinal fibrosis in Crohn’s disease through the ATX-LPA axis. J. Crohn’s Colitis 2022, 16, 1124–1139. [Google Scholar] [CrossRef]
- Yu, S.; Murph, M.M.; Lu, Y.; Liu, S.; Hall, H.S.; Liu, J.; Stephens, C.; Fang, X.; Mills, G.B. Lysophosphatidic acid receptors determine tumorigenicity and aggressiveness of ovarian cancer cells. JNCI J. Natl. Cancer Inst. 2008, 100, 1630–1642. [Google Scholar] [CrossRef]
- Schmitz, U.; Thömmes, K.; Beier, I.; Vetter, H. Lysophosphatidic acid stimulates p21-activated kinase in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2002, 291, 687–691. [Google Scholar] [CrossRef]
- Ara, H.; Subedi, U.; Sharma, P.; Bhattarai, S.; Sharma, S.; Manikandan, S.; Yu, X.; Bhuiyan, M.S.; Sun, H.; Miriyala, S.; et al. Alteration of Cellular Energy Metabolism through LPAR2-Axin2 Axis in Gastric Cancer. Biomolecules 2022, 12, 1805. [Google Scholar] [CrossRef]
- Sharma, S.; Patel, F.; Ara, H.; Bess, E.; Shum, A.; Bhattarai, S.; Subedi, U.; Bell, D.S.; Bhuiyan, M.S.; Sun, H.; et al. Rotenone-Induced 4-HNE Aggresome Formation and Degradation in HL-1 Cardiomyocytes: Role of Autophagy Flux. Int. J. Mol. Sci. 2022, 23, 4675. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subedi, U.; Manikandan, S.; Bhattarai, S.; Sharma, P.; Sharma, S.; Sun, H.; Miriyala, S.; Panchatcharam, M. The Autotaxin-LPA Axis Emerges as a Novel Regulator of Smooth Muscle Cell Phenotypic Modulation during Intimal Hyperplasia. Int. J. Mol. Sci. 2023, 24, 2913. https://doi.org/10.3390/ijms24032913
Subedi U, Manikandan S, Bhattarai S, Sharma P, Sharma S, Sun H, Miriyala S, Panchatcharam M. The Autotaxin-LPA Axis Emerges as a Novel Regulator of Smooth Muscle Cell Phenotypic Modulation during Intimal Hyperplasia. International Journal of Molecular Sciences. 2023; 24(3):2913. https://doi.org/10.3390/ijms24032913
Chicago/Turabian StyleSubedi, Utsab, Shrivats Manikandan, Susmita Bhattarai, Papori Sharma, Sudha Sharma, Hong Sun, Sumitra Miriyala, and Manikandan Panchatcharam. 2023. "The Autotaxin-LPA Axis Emerges as a Novel Regulator of Smooth Muscle Cell Phenotypic Modulation during Intimal Hyperplasia" International Journal of Molecular Sciences 24, no. 3: 2913. https://doi.org/10.3390/ijms24032913
APA StyleSubedi, U., Manikandan, S., Bhattarai, S., Sharma, P., Sharma, S., Sun, H., Miriyala, S., & Panchatcharam, M. (2023). The Autotaxin-LPA Axis Emerges as a Novel Regulator of Smooth Muscle Cell Phenotypic Modulation during Intimal Hyperplasia. International Journal of Molecular Sciences, 24(3), 2913. https://doi.org/10.3390/ijms24032913