1. Introduction
Cytotoxic CD8+ T cells play an essential role in adaptive immunity. They recognize targets via the major histocompatibility complex (MHC) class I. Recognition triggers a cascade of T cell activation and signaling, resulting in the release of effector molecules, such as lytic substances and cytokines, to destroy target cells or to enhance the inflammatory response. Neurons and other brain cells express MHC I molecules and are susceptible to targeting by CD8+ T cells [
1,
2]. Neural antigen-reactive cytotoxic CD8+ T cells contribute to neuronal dysfunction and degeneration in a variety of inflammatory central nervous system (CNS) disorders, such as multiple sclerosis (MS), neuromyelitis optica, and acute disseminated encephalomyelitis [
3]. MS is the most common of these diseases. Both myelinated white matter and gray matter [
4,
5,
6] are affected by this autoimmune attack. The mechanisms of gray matter inflammation and degeneration in MS in the context of T cell autoimmune attack are not well understood. Experimental autoimmune encephalomyelitis (EAE) is a commonly used animal model for MS. In EAE, neural damage is caused by infiltrating autoreactive T cells, which react to neural antigens, such as myelin oligodendrocyte glycoprotein (MOG) and myelin basic protein (MBP), and attack the myelin sheath of neurons, resulting in CNS inflammation, demyelination, and neurodegeneration [
7,
8,
9].
Both CD4+ and CD8+ T cells are found at lesion sites in MS patients and EAE animals [
10], but CD8+ T cells are the predominant T cell population in human MS lesions [
11]. Their role in autoimmune pathologic processes remains unclear. Cytotoxic CD8+ T cells are considered important effector cells that contribute to neuronal damage [
12,
13,
14], but they have been reported to be neuroprotective and to limit neuronal damage in EAE [
15,
16,
17,
18]. This suggests that CD8+ T cells may play a role in regulating the balance between neuroprotection and neural damage through their interaction with brain cells. Furthermore, the brain possesses defense mechanisms that support neuronal plasticity and protect against inflammation and neuronal damage. This complex defense mechanism involves glia cells and neurons that actively remove infiltrating immune cells or minimize neuronal damage by inducing synaptic loss [
19,
20,
21,
22,
23]. Regulatory T cells play an essential role in suppressing inflammation. Neurons are reported to be highly immune regulatory. They induce proliferation of activated CD4+ T cells and convert these encephalitogenic T cells into regulatory cells via interactions with T cells that inhibit EAE [
24]. The use of EAE models has contributed to our understanding of the pathology of MS, but much remains to be learned about how brain cells interact with infiltrating T cells in the context of neuroprotection and neurodestruction.
To study how neural antigen-reactive cytotoxic CD8+ T cells interact with brain cells in inflammation, we developed in vitro neuronal cell culture and ex vivo acute brain slice co-culture models with activated T cells, allowing effector T cells to interact with neuronal cells. To visualize CD8+ T cells and neurons, we used CD8-cre-τGFP mice and Thy1.2-HcRed transgenic mice whose cortical neurons express the far-red fluorescent protein HcRed1. We used different effector T cell preparations adapted to in vitro and ex vivo co-culture studies. In addition, we generated inflammatory conditions in these two models by supplying T cell conditioned medium that contained a variety of cytokines produced by CD8+ T cells during their polyclonal activation. We verified the inflamed condition in both co-culture models. Changes in T cell migration were observed by live-cell confocal imaging, and an increase in two key proinflammatory cytokines, interferon gamma (IFNγ) and TNF alpha (TNFα), was confirmed by ELISA assay. Our in vitro and ex vivo models provide an easily manipulated platform for studying the molecular details of neuron–immune cell interaction under inflammatory conditions. Combining these two models with high-resolution imaging techniques can potentially uncover molecular details of neuropathological processes or neuroprotective mechanisms that are difficult to extract from in vivo studies due to limited visualization access and the complexity of cell populations.
3. Discussion
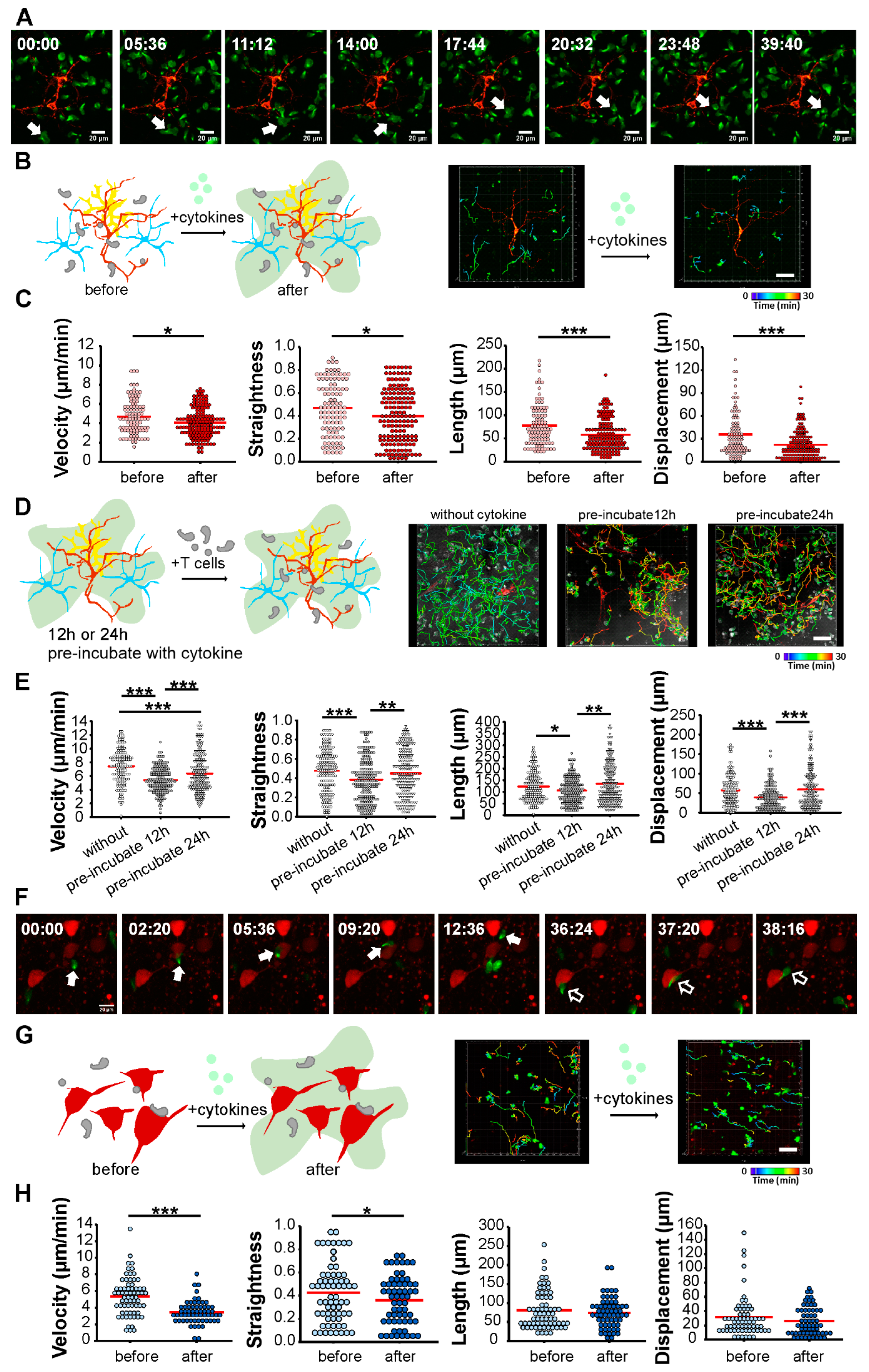
Multiple sclerosis is an autoimmune disease of the CNS. Autoreactive T cell infiltration into the CNS contributes to inflammation and subsequent pathology. This process can be mimicked by EAE, an animal model of MS. Different EAE models, including mouse strains, transferred encephalitogenic T cell clones, and transgenic recipient mouse lines, phenocopy different aspects of MS. In MS, lesions appear in both white and gray matter. To understand the cortical damage caused by autoreactive T cells, we established in vitro and ex vivo T cell–neuron inflammatory models and analyzed how T cells responded to the inflamed neuronal network. We visualized CD8+ T cells migrating on the neuronal network, as well as within neocortices, by live confocal imaging. Contact and interaction between T cells and cortical neurons can be observed in the in vitro and ex vivo models (
Figure 3A,F). In addition, reduced migration velocity was detected under inflamed conditions in both models (
Figure 3), consistent with in vivo observations [
31,
39].
Although T cell–neuron co-culture models have been used previously [
40,
41], here we applied systematic in vitro and ex vivo preparations to study the interactions of T cells with neurons during inflammation. These models are easily adaptable and manipulatable. For instance, as many infiltrating immune cell populations have been identified at the lesion sites of MS patients, one could investigate neuronal interactions with target immune cell types (i.e., B cells or NK cells) by applying them to neuron cultures or brain slices. To investigate specific inflammatory pathways affecting neurons or glia cells, one could control the cytokine concentration or cytokine cocktail combination to uncover the complexity of the inflammatory response by mimicking chronic or acute neuroinflammation. The models we provide here offer significant accessibility for analyzing and monitoring various cellular activity parameters of T cells and neuronal cells, such as changes in calcium signaling, synaptic transmission features, and T cell or glia cell activation under neuroinflammatory conditions. Immune cell activation plays a part in modulating the acute and chronic progression of neurodegenerative diseases [
42]. Despite the strength of the in vitro and ex vivo model combination, a lack of physiological integrity remains when studying neurological–autoimmune pathology. The unmodified crosstalk between the immune system and CNS is preserved solely in the living organism. Therefore, in vivo studies are still indispensable. It is important to verify the findings from in vitro and ex vivo studies using in vivo models.
Isolated brain cells from the neocortex of postnatal day 0 pups consisted mainly of cortical neurons. Due to a lack of oligodendrocytes, myelin was absent in our neuronal culture. Therefore, we generated polyclonal activated effector CD8+ T cells without priming them to neural antigens for the in vitro study. We optimized the co-culture conditions by using Neuronal-A culture medium due to the susceptibility of neurons. Activated CD8+ T cells appeared to tolerate the NBA medium well in the co-culture. For the ex vivo study, we generated neural antigen-reactive T cells by priming naive T cells from splenocytes with the MOG
35-55 peptide, the peptide clone that can induce EAE in C57BL/6 mice. When co-culturing ex vivo brain slices, we immersed the brain slices and T cells in standard artificial cerebrospinal fluid, which was also well tolerated by the T cells. The quality of the brain slice is decisive for fine neuronal activity measurement. We cut acute brain slices in ice-cold high sucrose buffer using a vibratome to minimize neuronal damage. The use of ice-cold buffer for brain slicing has been applied extensively for performing patch clamping [
43,
44,
45], the most sensitive method for measuring neuronal activity. Eguchi et al. compared ultrastructural and electrophysiological features of synapses in mouse acute brain slices prepared at ice-cold and physiological temperatures (35–37 °C). The results pointed out that preparation under physiological temperature provides substantial advantages for investigating synaptic functions [
46]. It might be worthwhile to test whether acute brain slices prepared under physiological temperature better preserve brain cellular activities, which consequently fine tune the regulation of the T cell attack in the co-culture system.
Recombinant proinflammatory cytokines, such as IL-1β, IFNγ, and TNFα, are often used to produce neuroinflammation [
40,
41,
43]. These cytokines generate an inflammatory response with changes in neuronal calcium signaling, electrical activity, and T cell migration. Inflammation consists of a tightly regulated process associated with changes in the cytokines produced by cells. It is unclear which cytokines occur at the site of pathology and what kind of effects they contribute locally and at other locations. To mimic the inflamed brain situation, instead of adding solely proinflammatory cytokines IFNγ and TNFα, we added conditioned medium produced by CD8+ T cells during their polyclonal activation. As autoreactive T cells undergo reactivation via contact with leptomeningeal phagocytes shortly before entering the CNS [
9], we bypassed this reactivation process by utilizing preactivated effector T cells to neuronal culture or brain slices.
We visualized real-time T cell migration on cortical neurons and in neocortical brain slices. Under inflamed conditions, CD8+ T cells drastically reduced their migration velocity and altered their migration pattern. This altered migration appeared in both the in vitro and ex vivo models (
Figure 3), which is in good agreement with the behavior of neural antigen-reactive infiltrating T cells in the inflamed brains of EAE animals [
31,
39]. Reduced T cell migration speed implies antigen recognition and cell activation through synapse formation. MHC I expression is upregulated in brain cells followed by IFNγ exposure [
1,
2], thereby increasing the likelihood that CD8+ T cells will recognize MHC I- expressing cells. Furthermore, CD8+ T cells formed synapses upon MHC I molecule recognition and attacked their cognate target cells [
44]; supplemented cytokines might support the activation of T cells. Although antigen-specific contacts of myelin protein-primed T cells to neurons contribute to reduced migration speed in contrast to antigen-unprimed T cells [
45], the supplemented cytokine appears to overcome the effects and change the behavior of effector T cells on neuronal cells and within neocortices (
Figure 3). Notably, MHC I molecules have been reported to play a role in synaptic plasticity and regeneration of neurons after nerve damage [
44,
46]. Since CD8+ T cells play a double-sided role in neuronal damage and neuroprotection, it would be interesting to know whether CD8+ T cells also contribute to neuroregeneration via MHC I engagement signaling.
Neuroinflammation can be intrinsic and lead to blood–brain barrier leakage and infiltration of T cells. In this scenario, autoreactive T cells enter the inflamed CNS before they contribute their proinflammatory cytokines. Glia cells, which maintain and support neuronal function, release a broad spectrum of inflammatory cytokines when challenged. However, brain cells react to the culture environment, and isolated glia cells tend to be activated in the cell culture, as well as in the sliced brain. Increased glial fibrillary acidic protein (GFAP) expression in astrocytes indicating astroglial activation was observed in the neuronal culture and acute brain slices (
Figure 1C), similar to that following brain injury or stress [
33]. We examined how CD8+ T cells responded in inflamed conditions and how long the supplemented cytokine effects remained in the neuronal culture. T cells showed dramatically reduced migration velocity in the 12 h pre-incubated neuronal culture (
Figure 3D,E), but their velocity recovered during the 24 h pre-incubation. We speculate that neuronal cells, in response to cytokines released from T cells, express MHC I or other inflammatory-driven upregulated molecules, such as intercellular adhesion molecule-1 (ICAM-1) [
47,
48]. ICAM-1 is the counter ligand of lymphocyte function-associated antigen-1 (LFA-1) [
49]. It is known to play essential roles in inflammatory processes and in T cell-mediated host defense mechanisms, resulting in decreased T cell migration velocity. After 24 h of inflammation, neurons may respond by reducing this response (e.g., by reducing MHC I molecule expression), resulting in a slight increase in T cell migration speed compared to controls. Overall, the migration data indicate that our in vitro and ex vivo models allowed for the study of T cell–neuronal interaction and the molecular basis of neuroinflammation in 2D and 3D neuronal networks.
Finally, our ELISA data verified production of the two key proinflammatory cytokines, IFNγ and TNFα, in both co-culture models within 24 h. It is surprising that when supplemented with cytokines in the absence of T cells, neuronal cells and brain slices are able to release high amounts of IFNγ and TNFα. Additional cytokines were produced when T cells were introduced. These data reveal the fact that activated T cells induced neuroinflammation through cytokine release and interaction with resident brain cells, which leads to synergistic effects on cytokine secretion. Moreover, the increases in cytokines were similar in the neuron culture and brain slices when co-cultured with T cells. IFNγ showed a linear increase, while TNFα showed an exponential increase in both co-cultured models (
Figure 4 and
Supplementary Figure S3). Astrocytes (62%) were the main population, followed by neurons (35%), in the in vitro culture, while neurons (72%) were the dominant cell population in the neocortices, followed by astrocytes (13%) (
Figure 1). We did not expect such consistency in these responses since microglia, the main TNFα producers, were notably underrepresented in the in vitro neuronal culture. Despite the differences in cell composition and neuronal network structure, it appears that neuron–glia networks can generate additional cytokine production, possibly to retain homeostasis against cell stress. The ELISA data revealed that our in vitro and ex vivo brain slice models responded in a similar fashion to neuroinflammation.
In this study, we established in vitro and ex vivo co-culture models mimicking neuroinflammatory conditions to study T cell and neuronal interaction. We verified the inflamed condition in both models by detecting the release of two key proinflammatory cytokines (IFNγ and TNFα). These inflamed neuronal networks resulted in a reduced T cell migration phenotype, which is consistent with the phenotype in the inflamed brains of EAE animals from other studies. Our in vitro and ex vivo models provide a promising platform for studying the molecular details of neuronal and immune cell interaction under inflammatory conditions, which is amenable to manipulation of numerous experimental factors.
4. Materials and Methods
4.1. Mice
Wild-type (WT) postnatal day 0 or day 1 pups were used for neuronal culture. The 2–3-week-old transgenic mice TgN (Thy1.2-HcRed)
THRE containing far-red fluorescent protein HcRed1-expressing neurons, driven by the Thy1.2 promoter [
50], were used to prepare the acute brain slices. The 8–20-week-old CD8cre1-τGFP mice were used to acquire T cells. CD8cre-τGFP mice were generated by crossing a previously published reporter τGFP line with a CD8a-Cre line (Jackson Laboratory, #008766). The WT mice and transgenic mice used in this study were all in C57BL/6N background. Both female and male mice were used in the experiments. The animals were kept under housing conditions of 22 °C room temperature (RT) with 50–60% humidity and 12 h dark/light cycles. All experimental procedures were approved and performed according to the regulations of the state of Saarland (Landesamt für Verbraucherschutz, AZ.: 2.4.1.1). All animal experiments were performed according to German law and European directives and with permission from the state of Saarland (Landesamt für Gesundheit und Verbraucherschutz; animal license number 41-2016, Perfusion (FKI/2020)).
4.2. Plasmid and Lentivirus Production
Neuron-specific infection pCDH-hSyn vectors were amplified from the backbone vector pZac2.1 hSynapsin1 NAPA-N2 SV40 (addgene #97212) using the forward primer 5′-ATGTATAGATATCAGTGCAAGTGGGTTTTAGGACCAG-3′ and reverse primer 5′- ATGTATACGCGGATCCCTGCGCTCTCAGGCACGACAC-3′ with restriction sites EcoRV and BamHI. The amplified hSyn product was then subcloned into a pCDH-EF1 (Addgene #72266) lentiviral transfer vector by replacing the elongation factor-1 alpha promoter (EF1α). The fluorescent protein mScarlet I was subcloned into a pCDH-hSyn vector with XbaI and XmaI restriction sites. The cDNA of the 4x mitochondrial targeting signal (4xmts) with the restriction sites NotI and PstI was purchased from the company BioCAT in pBluescript II SK(+) vector. The insert 4xmts cDNA was cloned into a pCDH-hSyn-mScarlet I vector. All constructs were verified with DNA sequence analysis (Microsynth Seqlab, Göttingen, Germany).
Lentivirus was produced using the packaging cell line Lenti-X293 (cat. 632180; Takara, Shiga, Japan) as described by the manufacturer. Briefly, 11 × 106 Lenti-X293 cells in a 10 cm dish were starved in DMEM medium without FCS supplement for 5 h before calcium phosphate transfection. Buffer (2x HBS) containing 280 mM NaCl, 50 mM HEPES, and 1.5 mM NaH2PO4 was added dropwise into a calcium-based plasmid mix solution (1.8 mL sigma water + 200 μL 2.5 M CaCl2 solution) containing 180 μg lentiviral-expressing plasmid (pCDH-hSyn-4xmts-mScarlet), 80 μg pMD2.G (Plasmid #12259; Addgene), 80 μg pMDLg/pRRE (Plasmid #12251; Addgene), and 80 μg pRSV-Rev (Plasmid #12253; Addgene). The plasmid mixed solution was incubated for 25 min at RT before application to the starved Lenti-X293 cells. The lentiviral particles were harvested 2.5 days after transfection. Finally, the viral particles were collected, followed by ultracentrifugation for 2.5 h at 22,500 rpm at 4 °C.
4.3. Acute Brain Slice Preparation
Thy1.2-HcRed mice were sacrificed by CO2 inhalation and decapitated. Their brains were quickly removed and placed into ice-cold high sucrose buffer containing (in mM): 230 Sucrose Ultra, 2 KCl, 1 KH2PO4, 10 D-(+)-glucose monohydrate, and 26 NaHCO3. Coronal brain slices (350 mm thick) were cut using a vibrating microtome (Leica VT1200 S) and transferred using a glass Pasteur pipette to a recovery chamber containing artificial cerebrospinal fluid (ACSF). The ACSF contained (in mM): 120 NaCl, 2 KCl, 1.3 MgCl2, 1 KH2PO4, 2 CaCl2, 26 NaHCO3, and 10 D-(+)-glucose monohydrate and was saturated with carbogen (95% O2 and 5% CO2 mixture; pH 7.4). The brain slices were recovered in the carbogenated ACSF at 35 °C for at least 2 h before the experiments.
4.4. Cell Culture
4.4.1. T Cell Culture for In Vitro Study
Splenocytes were isolated from the spleens of CD8cre-τGFP mice followed by smashing through a 70 µm cell strainer in cold RPMI medium supplemented with 10% FCS and 1% penicillin/streptomycin (P/S). The splenocytes were collected after removing erythrocytes from the cell suspension using a water-based lysis buffer containing 155 mM NH4Cl, 10 mM KHCO3, and 0.13 mM EDTA. An amount of 1 mL lysis buffer was applied to the splenocyte pellet from one spleen for 30 s at RT. Afterward, 9 mL RPMI medium was supplied to the 15 mL falcon to stop the lysis reaction. After centrifuging the cells at 300× g for 6 min, the cell suspension was further washed once with D-PBS containing 2 mM EDTA and 0.1% BSA before CD8 positive isolation. Naive CD8+ T cells were positively isolated from the splenocytes using a Dynabeads FlowComp Mouse CD8+ kit (Invitrogen) as described by the manufacturer. The isolated naive CD8+ T cells were stimulated with anti-CD3/anti-CD28 activator dynabeads (1:0.8 ratio; Invitrogen) and cultured for 5 days. The cells were cultured at a density of 1 × 106 cells/mL in a 24-well culture plate (2 million cells per well) with AIMV medium (Invitrogen) containing 10% FCS, 1% P/S (Invitrogen), and 50 μM 2-mercaptoethanol (BME). Afterward, 50 U/mL recombinant mouse IL-2 (Gibco) was applied in the first 2 days of T cell culture. Then, 100 U/mL mouse IL-2 was added after 48 h of culture to support T cell proliferation. These activated effector CTLs were used for the neuronal cell co-culture study.
4.4.2. MOG-Specific T Cell Culture
To generate MOG-specific T cells, naive splenocytes isolated from WT mice or CD8cre-τGFP transgenic mice were incubated with 75 µg/mL MOG35-55 peptide (Genaxxon) in RPMI culture medium containing 50 µM BME, 10% FCS, and 1% P/S that was supplemented with 0.5 µg/mL anti-CD3e (BD Pharmingen, clone 145-2C11, cat. 557306), 0.2 µg/mL anti-CD28 antibody (BD Pharmingen, cat. 553294), and 50 U/mL mouse IL-2 for 2 days. The splenocytes from one spleen were cultured in 5 mL of culture medium in one well of a 6-well plate. Blast cells were generated after 2 days (day 2) in culture. Then, 100 U/mL of IL-2 was added to the culture medium at day 3 to induce the proliferation of T cells. The T cells were separated from mixed splenocytes at day 3 or day 4 using Lymphocyte Separation Medium 1077 (PromoCell) with Ficoll density gradient centrifugation at 400× g for 30 min without break at RT. The purified T cells (1 × 106 cells/mL) were further cultured in RPMI culture medium supplemented with 100 U/mL IL-2 for 4 days (day 6). To expand the T cell population, we repeated the T cell priming protocol as with naive splenocytes but using thymocytes from another WT mouse with C57BL/6N background as antigen-presenting cells and restimulated the T cells on day 5 or day 6 with 75 µg/mL MOG peptide (second restimulation, day 0). It is notable that in the second cycle of stimulation, we did not provide suspended anti-CD3/anti-CD28 antibodies because these T cells were activated previously. The T cells formed blast cells after 2 days of priming. We utilized these cells after T cell purification with Ficoll density gradient centrifugation on day 4 for brain slice co-culture.
Thymocytes were isolated from the thymus by smashing through a 70 µm cell strainer in cold RPMI medium with 10% FCS. The thymocytes were collected to co-culture with day 5 or day 6 T cells (thymocyte:T cell ratio = 20:1) for restimulation. Five million T cells and 100 million thymocytes were co-cultured in one well of a 6-well plate in RPMI culture medium for 2 days, as in the stimulation protocol described above. Day 4 and day 5 MOG-specific T cells after thymocyte restimulation were used for cell migration studies with brain slices after lymphocyte purification.
4.4.3. Primary Cortical Neuron Culture
Cortical neurons were isolated from postnatal day 0–1 WT pups. Briefly, the cortices were isolated from the whole brain in ice-cold Earle’s Balanced Salt Solution (EBSS, Gibco). After peeling off the meninges, the cortices were digested with 35 units/mL papain (Worthington, NJ) for 45 min at 37 °C, followed by gentle mechanical trituration. Single cell suspensions were seeded on 25 mm glass coverslips placed in 6-well culture plates (3 × 105 cells per coverslip). The glass coverslips were precoated with a mixture of coating solution containing 17 mM acetic acid, poly-D-lysine (Sigma, St. Louis, MI, USA, P6407), and collagen I (Gibco, A1048301). The neurons were cultured in Neuronal-A (NBA) medium containing 10% FCS, 1% P/S, 1% GlutaMAX, and 2% B-27 supplement (Gibco) for 8–14 days before the experiments. The culture medium was replaced with fresh medium on the second day (day 2) to remove the remaining cell debris from the cell preparation. Afterward, neurons stayed in the conditional medium throughout the experiments, unless additional supplements are mentioned. To label mitochondria in the neurons, the neuronal cells were infected with lentivirus for 20 h in NBA medium without antibiotics on the next day of cell seeding. After 20 h, the virus was removed by replacing with fresh NBA culture medium. The neurons started to express mitochondrial markers 5 days after infection.
4.4.4. T Cell–Neuron Co-Culture for In Vitro Study
Day 3–4 bead-activated CD8+ T cells were used to co-culture with up to 8-day-old neuronal cells. The T cells (0.5 × 106) were added to each neuronal culture well containing 2 mL NBA culture medium for co-culture. To induce an inflamed condition, 50 µL T cell conditioned medium was added to each neuronal coverslip containing 2 mL of NBA medium.
4.4.5. T Cell–Brain Slice Co-Culture for Ex Vivo Study
Coronal brain slices were further cut sagittally into half pieces before co-culturing with T cells. MOG-specific T cells (2 × 106) were applied to one half brain slice in ACSF. Four half brain slices were placed in one well of a 6-well plate with a total of 8 × 106 T cells per well. To induce an inflamed condition, 150 μL of T cell conditioned medium was added to each well containing 2 mL of ACSF.
Co-cultured conditions were used for the T cell migration assay as well as ELISA analysis. Further technical details will be described in the sections below. All the cell cultures were controlled at 37 °C with 5% CO2, and brain slices were carbogenated with a 95% O2 and 5% CO2 mixture at 35 °C.
4.5. Confocal Imaging
For the live T cell migration in vitro assay, day 3 or day 4 activated CD8-τGFP T cells were applied to an imaging chamber containing neuronal cells in 2 mL NBA conditional culture medium (before cytokine application). T cell migration was recorded using a confocal microscope (LSM 780, Zeiss, Germany). After 45 min of recording, 50 μL of T cell conditioned medium (cytokine cocktail) was applied to mimic inflamed conditions. Afterward, another 45 min was recorded (after cytokine application). For T cell migration on brain slices (ex vivo assay), image recording was first started 2 h after the MOG-specific T cells were applied to the brain slices in order to visualize penetrating T cells in the brain slices. An amount of 150 µL T cell supernatant was applied to four pieces of half-brain slices in 2 mL ACSF to create the inflamed condition. Live imaging was performed at 37 °C for the in vitro assay or at 35 °C for the ex vivo assay with a stable carbogen supply (95% O2 and 5% CO2 gas mixture). Image acquisition information is stated in each figure, including z-stack and time resolution. The images were acquired with a 40x Plan-Apochromat objective (NA 1.4). For live and fixed samples, τGFP and anti-NeuN-Alexa488 fluorescence was excited at a 488 nm excitation wavelength, anti-GFAP-Alexa568 was excited at 568 nm, and anti-Iba1-Alexa647 was excited at 647 nm. For the brain slices, the image stacks were acquired as 12 µm total thickness with a 2 µm interval distance. For the cultured cells, 6 µm total thickness with a 1 µm interval distance was acquired. The maximum intensity projection images are shown in the live-cell imaging data.
4.6. ELISA
For ELISA analysis, supernatants were collected after 10 min, 6 h, 12 h, 18 h, and 24 h from T cell–neuron co-culture or T cell–brain slice co-culture. The concentrations of IFNγ and TNFα were quantitatively measured using an IFNγ ELISA kit (abcam, ab46081) and a TNFα ELISA kit (abcam, ab208348) as described by the manufacturer. Briefly, 100 μL of 1:10 diluted culture supernatants or 50 μL of 1:2 diluted culture supernatants were added to the IFNγ antibody or TNFα antibody precoated 96-well plates for 1 h, respectively, to form an antibody–antigen complex. After the washing steps, 100 μL TMB substrate was applied to the wells for 30 min at RT in the dark. Sequentially, 50 μL of stop solution was added to stop the reaction. Finally, the samples were read (TECAN; infinite M200 pro plate reader) at 450 nm wavelength. Four independent co-culture preparations were performed in both cytokine measurements.
4.7. Immunofluorescence
For brain slice staining, p12 WT mice were deeply anesthetized using an intraperitoneal injection of a mixture of ketamine (280 mg/kg bodyweight) and xylazine (20 mg/kg bodyweight). The mice were intracardially perfused with D-PBS for 5 min, followed by a fixative containing 4% paraformaldehyde for another 5 min. After perfusion, the brains were isolated and post-fixed in the same fixative for another 24 h. After washing once with PBS, the brains were sliced into 50 µm sections using a vibratome. The fixed brain slices were washed once with PBS and permeabilized with 0.1% triton (permeabilization buffer) for 10 min, followed by blocking with an extra 10% FCS in permeabilization buffer for 30 min at RT. Afterward, the samples were incubated overnight at 4 °C with primary antibodies anti-NeuN (1:200; Merck, clone A60), anti-GFAP (1:200; Abcam, ab4674), and anti-Iba1 (1:200; Wako, Code No. 019-19741). After washing three times with permeabilization buffer, corresponding secondary Alexa fluorophore-coupled antibodies were further incubated with the brain slices for 1 h at RT. Finally, the samples were washed three times with D-PBS, followed by DAPI (1:1000) staining and mounted with Fluoromount-G Mounting Medium (Invitrogen). For neuronal cell staining, the cells were fixed on day 12 with 4% PFA for 10 min at RT. After washing three times with D-PBS, the cells were stained following the staining protocol as for the brain slices except using 2% BSA for blocking.
For the MOG-specific lymphocyte staining shown in
Figure 2C, second stimulated day 4 cells (0.2 × 10
6) isolated from WT mice were collected from the culture in 1.5 mL Eppendorf tubes. The cells were resuspended in D-PBS and stained with anti-CD4-APC (1:200; eBioscience, GK1.5) and anti-CD8a-PE-CF594 (1:200; BD Horizon, Clone 53-6.7) antibodies for 45 min on ice. Afterward, the cells were spun down with 300
g for 5 min and washed once with 1 mL of D-PBS to remove unbound antibodies. Finally, the cells were resuspended again in 300 μL of D-PBS for confocal imaging.
4.8. Statistics and Image Analyses
The Mann–Whitney U test was used to compare values for T cell migration analysis. Values were considered statistically significant when probability (P) values were below 0.05 (*), 0.01 (**), or 0.001 (***). Data were analyzed with ImageJ v1.46 [
51], Excel (part of Office 2013, Microsoft), SigmaPlot 14, and Imaris 9.3 (Bitplane AG) and graphed using Affinity Designer Software (Serif Ltd.). Fitting analysis for IFNγ and TNFα release as a function of time was performed with SigmaPlot 14 using linear regression and a single exponential growth function.
For T cell migration analysis, confocal live images were exported to Imaris software to analyze CD8+ T cell migration co-cultured with neuronal cells. T cell movements were tracked using the Imaris spot object-based algorithm on source channel 488 to create tracks for each cell. Due to the uneven cell migration duration and tracks in the field of view, we included only the tracks dwelling over 15 min of the recordings in order to acquire a more accurate mean migration speed. All cell tracks were manually corrected using the manual editor when misdetection, nondetection, or false cell duplicates were observed. Some smaller debris or dead cells were detected by the software, despite the use of a threshold of 5–6 uM. Additionally, overlapping cells resulting in false single-track detection were manually corrected. Overall, with background subtraction and the set threshold mentioned above, we set the quality threshold for CD8+ T cell selection to 15–20% in our analysis. The parameters collected from migration analysis were defined as follows: Track duration (min): duration between the first and last time point within the tracks; Track length (μm): total length of the measured track; Track speed/velocity mean (μm/min): track length divided by the time between the first and last object within the track; Displacement (μm): straight distance between the first and last cell positions; and Track straightness: displacement divided by track length.



{kind=link}
{kind=link}
{kind=link}
{kind=link}



