

In Silico Analysis of the Structural Dynamics and Substrate Recognition Determinants of the Human Mitochondrial Carnitine/Acylcarnitine SLC25A20 Transporter




Abstract
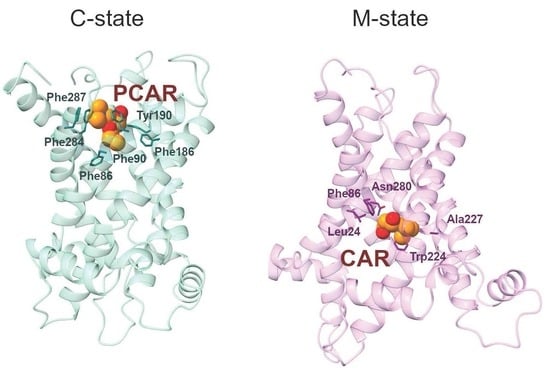
:
1. Introduction
2. Results and Discussion
2.1. Analysis of the Cytoplasmic and Matrix State Structural Models
2.2. Structural Asymmetry and Intraprotein Interactions
2.2.1. TM Helices Interfaces and Geometry: C- and M-State Comparison
2.2.2. C-State Intraprotein Interactions
2.2.3. M-State Intraprotein Interactions
2.2.4. Interactions Involving Substrate Contact Point Residues
2.3. Substrates Early Recognition Step
2.3.1. Phe287 and Tyr190 Are Involved in Acylcarnitine Early Recognition
2.3.2. Trp224 and Asn280 Are Involved in Carnitine Early Recognition
3. Materials and Methods
3.1. Sequence Analysis
3.2. Protein Structure Prediction
3.3. Molecular Dynamics Simulation
3.4. MD Simulation Analyses
3.5. Clustering Procedure
3.6. Molecular Docking
3.7. MD Simulation of the Protein-Ligand Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F.; Pierri, C.L. Structure and Function of Mitochondrial Carriers—Role of the Transmembrane Helix P and G Residues in the Gating and Transport Mechanism. FEBS Lett. 2010, 584, 1931–1939. [Google Scholar] [CrossRef] [Green Version]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.-M.; Brandolin, G. Structure of Mitochondrial ADP/ATP Carrier in Complex with Carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, A.J.; Kunji, E.R.S. Mitochondrial Carriers in the Cytoplasmic State Have a Common Substrate Binding Site. Proc. Natl. Acad. Sci. USA 2006, 103, 2617–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonazzi, A.; Giangregorio, N.; Console, L.; Palmieri, F.; Indiveri, C. The Mitochondrial Carnitine Acyl-Carnitine Carrier (SLC25A20): Molecular Mechanisms of Transport, Role in Redox Sensing and Interaction with Drugs. Biomolecules 2021, 11, 521. [Google Scholar] [CrossRef] [PubMed]
- Indiveri, C.; Iacobazzi, V.; Tonazzi, A.; Giangregorio, N.; Infantino, V.; Convertini, P.; Console, L.; Palmieri, F. The Mitochondrial Carnitine/Acylcarnitine Carrier: Function, Structure and Physiopathology. Mol. Asp. Med. 2011, 32, 223–233. [Google Scholar] [CrossRef]
- Casals, N.; Zammit, V.; Herrero, L.; Fadó, R.; Rodríguez-Rodríguez, R.; Serra, D. Carnitine Palmitoyltransferase 1C: From Cognition to Cancer. Prog. Lipid Res. 2016, 61, 134–148. [Google Scholar] [CrossRef] [Green Version]
- De Pinto, V. Renaissance of VDAC: New Insights on a Protein Family at the Interface between Mitochondria and Cytosol. Biomolecules 2021, 11, 107. [Google Scholar] [CrossRef]
- Console, L.; Giangregorio, N.; Indiveri, C.; Tonazzi, A. Carnitine/Acylcarnitine Translocase and Carnitine Palmitoyltransferase 2 Form a Complex in the Inner Mitochondrial Membrane. Mol. Cell. Biochem. 2014, 394, 307–314. [Google Scholar] [CrossRef]
- Joshi, P.R.; Zierz, S. Muscle Carnitine Palmitoyltransferase II (CPT II) Deficiency: A Conceptual Approach. Molecules 2020, 25, 1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhao, F.; Zhao, Z.; Zhao, X.; Meng, H.; Zhang, D.; Zhao, S.; Ding, M. Neonatal Sudden Death Caused by a Novel Heterozygous Mutation in SLC25A20 Gene: A Case Report and Brief Literature Review. Leg. Med. 2022, 54, 101990. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Mu, J.; Wang, Z.; Ma, S.; Da, X.; Song, J.; Zhang, H.; Yang, L.; Li, J.; Yang, J. Down-Regulation of SLC25A20 Promotes Hepatocellular Carcinoma Growth and Metastasis through Suppression of Fatty-Acid Oxidation. Cell Death Dis. 2021, 12, 361. [Google Scholar] [CrossRef] [PubMed]
- Andersen, V.; Halekoh, U.; Tjønneland, A.; Vogel, U.; Kopp, T.I. Intake of Red and Processed Meat, Use of Non-Steroid Anti-Inflammatory Drugs, Genetic Variants and Risk of Colorectal Cancer: A Prospective Study of the Danish “Diet, Cancer and Health” Cohort. Int. J. Mol. Sci. 2019, 20, 1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falconi, M.; Chillemi, G.; Di Marino, D.; D’Annessa, I.; Morozzo della Rocca, B.; Palmieri, L.; Desideri, A. Structural Dynamics of the Mitochondrial ADP/ATP Carrier Revealed by Molecular Dynamics Simulation Studies. Proteins Struct. Funct. Bioinform. 2006, 65, 681–691. [Google Scholar] [CrossRef]
- Dehez, F.; Pebay-Peyroula, E.; Chipot, C. Binding of ADP in the Mitochondrial ADP/ATP Carrier Is Driven by an Electrostatic Funnel. J. Am. Chem. Soc. 2008, 130, 12725–12733. [Google Scholar] [CrossRef]
- Wang, Y.; Tajkhorshid, E. Electrostatic Funneling of Substrate in Mitochondrial Inner Membrane Carriers. Proc. Natl. Acad. Sci. USA 2008, 105, 9598–9603. [Google Scholar] [CrossRef] [Green Version]
- Pietropaolo, A.; Pierri, C.L.; Palmieri, F.; Klingenberg, M. The Switching Mechanism of the Mitochondrial ADP/ATP Carrier Explored by Free-Energy Landscapes. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 772–781. [Google Scholar] [CrossRef]
- Tamura, K.; Hayashi, S. Atomistic Modeling of Alternating Access of a Mitochondrial ADP/ATP Membrane Transporter with Molecular Simulations. PLoS ONE 2017, 12, e0181489. [Google Scholar] [CrossRef]
- Yi, Q.; Li, Q.; Yao, S.; Chen, Y.; Guan, M.-X.; Cang, X. Molecular Dynamics Simulations on Apo ADP/ATP Carrier Shed New Lights on the Featured Motif of the Mitochondrial Carriers. Mitochondrion 2019, 47, 94–102. [Google Scholar] [CrossRef]
- Mao, X.; Yao, S.; Yi, Q.; Xu, Z.M.; Cang, X. Function-Related Asymmetry of the Specific Cardiolipin Binding Sites on the Mitochondrial ADP/ATP Carrier. Biochim. Et Biophys. Acta-Biomembr. 2021, 1863, 183466. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Yi, Q.; Ma, B.; Mao, X.; Chen, Y.; Guan, M.-X.; Cang, X. Mechanistic Insights into Multiple-Step Transport of Mitochondrial ADP/ATP Carrier. Comput. Struct. Biotechnol. J. 2022, 20, 1829–1840. [Google Scholar] [CrossRef] [PubMed]
- Pasquadibisceglie, A.; Polticelli, F. Computational Studies of the Mitochondrial Carrier Family SLC25. Present Status and Future Perspectives. Bio-Algorithms Med-Syst. 2021, 17, 65–78. [Google Scholar] [CrossRef]
- Seccia, R.; De Santis, S.; Di Noia, M.A.; Palmieri, F.; Miniero, D.V.; Marmo, R.; Paradies, E.; Santoro, A.; Pierri, C.L.; Palmieri, L.; et al. Citrate Regulates the Saccharomyces Cerevisiae Mitochondrial GDP/GTP Carrier (Ggc1p) by Triggering Unidirectional Transport of GTP. J. Fungi 2022, 8, 795. [Google Scholar] [CrossRef] [PubMed]
- Vozza, A.; De Leonardis, F.; Paradies, E.; De Grassi, A.; Pierri, C.L.; Parisi, G.; Marobbio, C.M.T.; Lasorsa, F.M.; Muto, L.; Capobianco, L.; et al. Biochemical Characterization of a New Mitochondrial Transporter of Dephosphocoenzyme A in Drosophila Melanogaster. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Remani, S.; Sun, J.; Kotaria, R.; Mayor, J.A.; Walters, D.E.; Kaplan, R.S. Identification of the Substrate Binding Sites within the Yeast Mitochondrial Citrate Transport Protein. J. Biol. Chem. 2007, 282, 17210–17220. [Google Scholar] [CrossRef] [Green Version]
- Mavridou, V.; King, M.S.; Tavoulari, S.; Ruprecht, J.J.; Palmer, S.M.; Kunji, E.R.S. Substrate Binding in the Mitochondrial ADP/ATP Carrier Is a Step-Wise Process Guiding the Structural Changes in the Transport Cycle. Nat. Commun. 2022, 13, 3585. [Google Scholar] [CrossRef]
- del Alamo, D.; Sala, D.; Mchaourab, H.S.; Meiler, J. Sampling Alternative Conformational States of Transporters and Receptors with AlphaFold2. eLife 2022, 11, e75751. [Google Scholar] [CrossRef]
- Pierri, C.L.; Palmieri, F.; De Grassi, A. Single-Nucleotide Evolution Quantifies the Importance of Each Site along the Structure of Mitochondrial Carriers. Cell. Mol. Life Sci. 2014, 71, 349–364. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Indiveri, C.; Palmieri, F. Site-Directed Mutagenesis of Charged Amino Acids of the Human Mitochondrial Carnitine/Acylcarnitine Carrier: Insight into the Molecular Mechanism of Transport. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Tonazzi, A.; Console, L.; Giangregorio, N.; Indiveri, C.; Palmieri, F. Identification by Site-Directed Mutagenesis of a Hydrophobic Binding Site of the Mitochondrial Carnitine/Acylcarnitine Carrier Involved in the Interaction with Acyl Groups. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2012, 1817, 697–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayikci, M.; Venkatakrishnan, A.J.; Scott-Brown, J.; Ravarani, C.N.J.; Flock, T.; Babu, M.M. Visualization and Analysis of Non-Covalent Contacts Using the Protein Contacts Atlas. Nat. Struct. Mol. Biol. 2018, 25, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for Integrated Sequence-Structure Analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruprecht, J.J.; Hellawell, A.M.; Harding, M.; Crichton, P.G.; McCoy, A.J.; Kunji, E.R.S. Structures of Yeast Mitochondrial ADP/ATP Carriers Support a Domain-Based Alternating-Access Transport Mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, E426–E434. [Google Scholar] [CrossRef] [Green Version]
- Brüschweiler, S.; Yang, Q.; Run, C.; Chou, J.J. Substrate-Modulated ADP/ATP-Transporter Dynamics Revealed by NMR Relaxation Dispersion. Nat. Struct. Mol. Biol. 2015, 22, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Sounier, R.; Bellot, G.; Chou, J.J. Mapping Conformational Heterogeneity of Mitochondrial Nucleotide Transporter in Uninhibited States. Angew. Chem. Int. Ed. 2015, 54, 2436–2441. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Ma, B.; Yi, Q.; Guan, M.-X.; Cang, X. Investigating the Broad Matrix-Gate Network in the Mitochondrial ADP/ATP Carrier through Molecular Dynamics Simulations. Molecules 2022, 27, 1071. [Google Scholar] [CrossRef]
- Yi, Q.; Yao, S.; Ma, B.; Cang, X. Function-Related Asymmetry of the Interactions between Matrix Loops and Conserved Sequence Motifs in the Mitochondrial ADP/ATP Carrier. Int. J. Mol. Sci. 2022, 23, 10877. [Google Scholar] [CrossRef]
- Yi, Q.; Yao, S.; Ma, B.; Cang, X. The Effects of Cardiolipin on the Structural Dynamics of the Mitochondrial ADP/ATP Carrier in Its Cytosol-Open State. J. Lipid Res. 2022, 63, 100227. [Google Scholar] [CrossRef]
- Johnston, J.M.; Johnston, J.M.; Khalid, S.; Johnston, J.M.; Khalid, S.; Sansom, M.S.P. Conformational Dynamics of the Mitochondrial ADP/ATP Carrier: A Simulation Study. Mol. Membr. Biol. 2008, 25, 506–517. [Google Scholar] [CrossRef]
- Giangregorio, N.; Pierri, C.L.; Tonazzi, A.; Incampo, G.; Tragni, V.; De Grassi, A.; Indiveri, C. Proline/Glycine Residues of the PG-Levels Guide Conformational Changes along the Transport Cycle in the Mitochondrial Carnitine/Acylcarnitine Carrier (SLC25A20). Int. J. Biol. Macromol. 2022, 221, 1453–1465. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Invernizzi, F.; Baratta, S.; Pons, R.; Chung, W.; Garavaglia, B.; Dionisi-Vici, C.; Ribes, A.; Parini, R.; Huertas, M.D.; et al. Molecular and Functional Analysis of SLC25A20 Mutations Causing Carnitine-Acylcarnitine Translocase Deficiency. Hum. Mutat. 2004, 24, 312–320. [Google Scholar] [CrossRef]
- Giangregorio, N.; Console, L.; Tonazzi, A.; Palmieri, F.; Indiveri, C. Identification of Amino Acid Residues Underlying the Antiport Mechanism of the Mitochondrial Carnitine/Acylcarnitine Carrier by Site-Directed Mutagenesis and Chemical Labeling. Biochemistry 2014, 53, 6924–6933. [Google Scholar] [CrossRef] [PubMed]
- Pasquadibisceglie, A.; Polticelli, F. Structural Determinants of Ligands Recognition by the Human Mitochondrial Basic Amino Acids Transporter SLC25A29. Insights from Molecular Dynamics Simulations of the c-State. Comput. Struct. Biotechnol. J. 2021, 19, 5600–5612. [Google Scholar] [CrossRef]
- De Lucas, J.R.; Domínguez, A.I.; Valenciano, S.; Turner, G.; Laborda, F. The AcuH Gene of Aspergillus Nidulans, Required for Growth on Acetate and Long-Chain Fatty Acids, Encodes a Putative Homologue of the Mammalian Carnitine/Acylcarnitine Carrier. Arch. Microbiol. 1999, 171, 386–396. [Google Scholar] [CrossRef] [PubMed]
- De Lucas, J.R.; De Lucas, J.R.; Indiveri, C.; De Lucas, J.R.; Indiveri, C.; Tonazzi, A.; Perez, P.; Giangregorio, N.; Iacobazzi, V.; Palmieri, F. Functional Characterization of Residues within the Carnitine/Acylcarnitine Translocase RX2PANAAXF Distinct Motif. Mol. Membr. Biol. 2008, 25, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Pistillo, M.; Scalera, V.; Indiveri, C. Tryptophan 224 of the Rat Mitochondrial Carnitine/Acylcarnitine Carrier Is Crucial for the Antiport Mechanism. Biochim. Et Biophys. Acta-Bioenerg. 2019, 1860, 708–716. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Giangregorio, N.; Palmieri, F. Bacterial Overexpression, Purification, and Reconstitution of the Carnitine/Acylcarnitine Carrier from Rat Liver Mitochondria. Biochem. Biophys. Res. Commun. 1998, 249, 589–594. [Google Scholar] [CrossRef]
- Robinson, A.J.; Overy, C.; Kunji, E.R.S. The Mechanism of Transport by Mitochondrial Carriers Based on Analysis of Symmetry. Proc. Natl. Acad. Sci. USA 2008, 105, 17766–17771. [Google Scholar] [CrossRef] [Green Version]
- Monné, M.; Miniero, D.V.; Daddabbo, L.; Palmieri, L.; Porcelli, V.; Palmieri, F. Mitochondrial Transporters for Ornithine and Related Amino Acids: A Review. Amino Acids 2015, 47, 1763–1777. [Google Scholar] [CrossRef]
- Palmieri, F.; Pierri, C.L.; De Grassi, A.; Nunes-Nesi, A.; Fernie, A.R. Evolution, Structure and Function of Mitochondrial Carriers: A Review with New Insights. Plant J. 2011, 66, 161–181. [Google Scholar] [CrossRef]
- Vaca Jacome, A.S.; Rabilloud, T.; Schaeffer-Reiss, C.; Rompais, M.; Ayoub, D.; Lane, L.; Bairoch, A.; Van Dorsselaer, A.; Carapito, C. N-Terminome Analysis of the Human Mitochondrial Proteome. Proteomics 2015, 15, 2519–2524. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2--a Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Pasquadibisceglie, A.; Leccese, A.; Polticelli, F. A computational study of the structure and function of human Zrt and Irt-like proteins metal transporters: An elevator-type transport mechanism predicted by AlphaFold2. Front. Chem. 2022, 10, 1004815. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-Align: A Protein Structure Alignment Algorithm Based on the TM-Score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and Upgrading Automated Preparation of Biomolecular Structures for Molecular Simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical PKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Pogozheva, I.D.; Armstrong, G.A.; Kong, L.; Hartnagel, T.J.; Carpino, C.A.; Gee, S.E.; Picarello, D.M.; Rubin, A.S.; Lee, J.; Park, S.; et al. Comparative Molecular Dynamics Simulation Studies of Realistic Eukaryotic, Prokaryotic, and Archaeal Membranes. J. Chem. Inf. Model. 2022, 62, 1036–1051. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.I.; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. Amber22 2022. Available online: https://ambermd.org/AmberTools.php (accessed on 1 February 2023).
- Chow, K.-H.; Ferguson, D.M. Isothermal-Isobaric Molecular Dynamics Simulations with Monte Carlo Volume Sampling. Comput. Phys. Commun. 1995, 91, 283–289. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Bansal, M.; Kumart, S.; Velavan, R. HELANAL: A Program to Characterize Helix Geometry in Proteins. J. Biomol. Struct. Dyn. 2000, 17, 811–819. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 56–61.
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TM Helix | Residues Range | TM Helix | Residues Range | Total Atomic Contacts | |
|---|---|---|---|---|---|
| c-state | H1 | 13–29 | H2 | 78–88 | 269 |
| H2 | 77–100 | H3 | 112–135 | 462 | |
| H3 | 110–132 | H4 | 172–195 | 582 | |
| H4 | 174–197 | H5 | 211–238 | 759 | |
| H5 | 209–228 | H6 | 269–285 | 323 | |
| H6 | 270–295 | H1 | 12–38 | 714 | |
| m-state | H1 | 10–29 | H2 | 78–98 | 524 |
| H2 | 85–100 | H3 | 112–128 | 415 | |
| H3 | 110–132 | H4 | 172–195 | 551 | |
| H4 | 174–197 | H5 | 211–238 | 756 | |
| H5 | 209–228 | H6 | 269–288 | 541 | |
| H6 | 275–295 | H1 | 12–32 | 505 |
| MD1 | |||
|---|---|---|---|
| Residue 1 | Residue 2 | Fraction | Avg Distance (Å) |
| Asp231 | Lys35 | 0.94 | 2.9 |
| Asp32 | Arg275 | 0.84 | 3.0 |
| Glu132 | Lys234 | 0.50 | 2.8 |
| Asp32 | Lys135 | 0.20 | 2.8 |
| MD2 | |||
| Residue 1 | Residue 2 | Fraction | Avg Distance (Å) |
| Asp32 | Arg275 | 0.75 | 3.0 |
| Asp32 | Lys135 | 0.48 | 2.9 |
| Asp231 | Lys35 | 0.36 | 2.9 |
| Glu132 | Lys234 | 0.28 | 3.1 |
| MD1 | |||
|---|---|---|---|
| Residue 1 | Residue 2 | Fraction | Avg Distance (Å) |
| Asp32 | Arg275 | 0.94 | 3.0 |
| Glu288 | Lys194 | 0.78 | 2.9 |
| Glu288 | Tyr190 | 0.61 | 2.7 |
| Glu132 | Lys234 | 0.57 | 3.0 |
| MD2 | |||
| Residue 1 | Residue 2 | Fraction | Avg Distance (Å) |
| Asp32 | Arg275 | 0.84 | 3.0 |
| Glu288 | Lys194 | 0.70 | 3.0 |
| Glu132 | Lys234 | 0.51 | 3.0 |
| Glu288 | Tyr190 | 0.46 | 2.7 |
| MD1 | ||||
|---|---|---|---|---|
| Residue 1 | Residue 2 | Fraction | Avg Distance (Å) | Avg Angle (°) |
| Phe287 | PCAR | 0.73 | 4.8 | 88.7 |
| PCAR | Tyr190 | 0.39 | 2.7 | 163.7 |
| MD2 | ||||
| Residue 1 | Residue 2 | Fraction | Avg Distance (Å) | Avg Angle (°) |
| Phe287 | PCAR | 0.31 | 4.9 | 85.4 |
| PCAR | Tyr190 | 0.33 | 2.7 | 164.3 |
| MD1 | ||||
|---|---|---|---|---|
| Residue 1 | Residue 2 | Fraction | Avg Distance (Å) | Avg Angle (°) |
| Trp224 | CAR | 0.79 | 4.6 | 93.2 |
| CAR | Asn280 | 0.17 | 2.8 | 160.8 |
| MD2 | ||||
| Residue 1 | Residue 2 | Fraction | Avg Distance (Å) | Avg Angle (°) |
| Trp224 | CAR | 0.61 | 4.4 | 90.7 |
| CAR | Asn280 | 0.51 | 2.8 | 160.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquadibisceglie, A.; Quadrotta, V.; Polticelli, F. In Silico Analysis of the Structural Dynamics and Substrate Recognition Determinants of the Human Mitochondrial Carnitine/Acylcarnitine SLC25A20 Transporter. Int. J. Mol. Sci. 2023, 24, 3946. https://doi.org/10.3390/ijms24043946
Pasquadibisceglie A, Quadrotta V, Polticelli F. In Silico Analysis of the Structural Dynamics and Substrate Recognition Determinants of the Human Mitochondrial Carnitine/Acylcarnitine SLC25A20 Transporter. International Journal of Molecular Sciences. 2023; 24(4):3946. https://doi.org/10.3390/ijms24043946
Chicago/Turabian StylePasquadibisceglie, Andrea, Virginia Quadrotta, and Fabio Polticelli. 2023. "In Silico Analysis of the Structural Dynamics and Substrate Recognition Determinants of the Human Mitochondrial Carnitine/Acylcarnitine SLC25A20 Transporter" International Journal of Molecular Sciences 24, no. 4: 3946. https://doi.org/10.3390/ijms24043946
APA StylePasquadibisceglie, A., Quadrotta, V., & Polticelli, F. (2023). In Silico Analysis of the Structural Dynamics and Substrate Recognition Determinants of the Human Mitochondrial Carnitine/Acylcarnitine SLC25A20 Transporter. International Journal of Molecular Sciences, 24(4), 3946. https://doi.org/10.3390/ijms24043946