
Role of UCP2 in the Energy Metabolism of the Cancer Cell Line A549

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
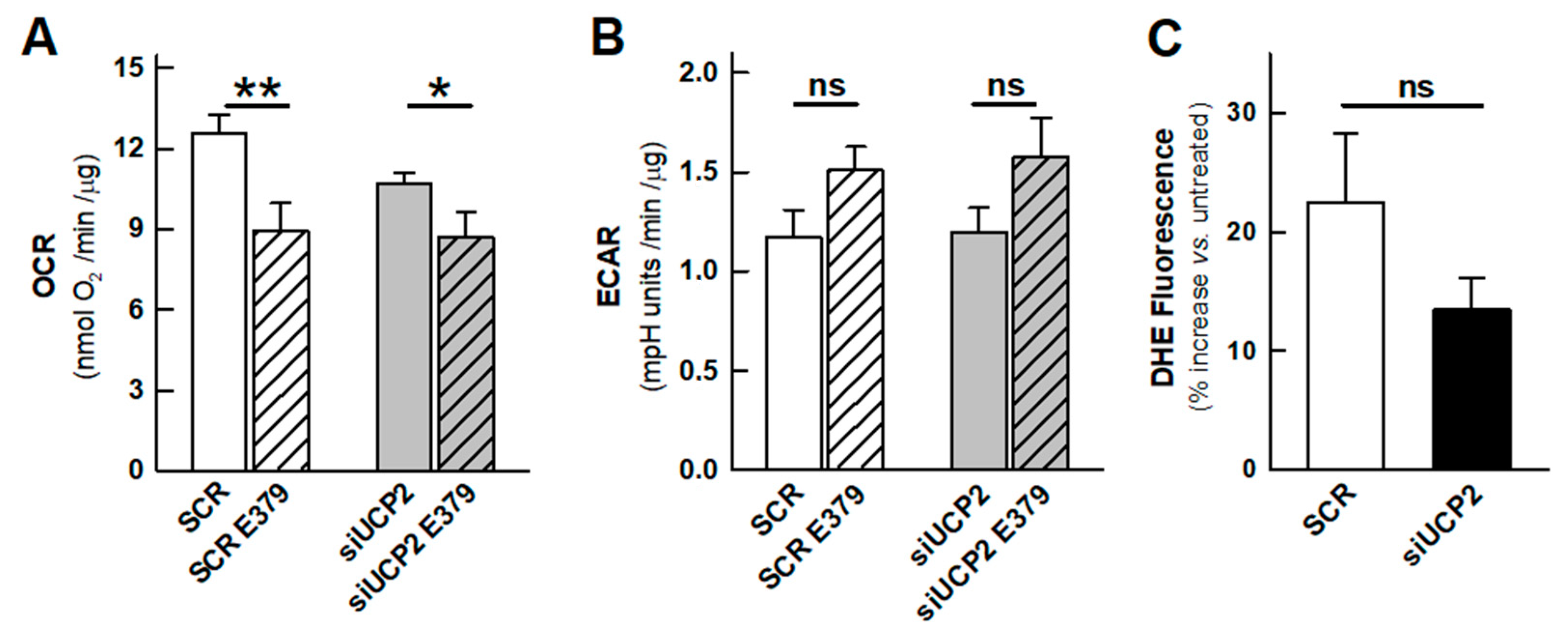
2.1. OXPHOS and Aerobic Glycolysis
2.2. Reactive Oxygen Species
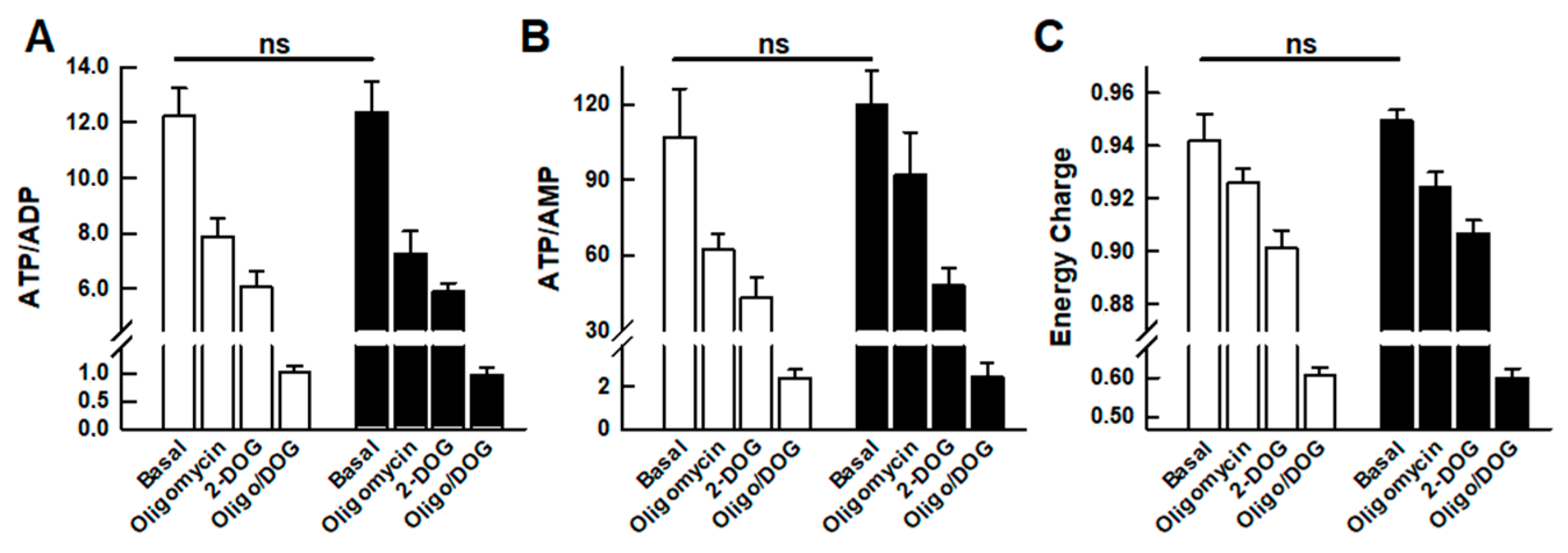
2.3. Adenine Nucleotide Levels
2.4. Cell Growth
2.5. Chromane Derivatives
3. Discussion

4. Materials and Methods
4.1. Cell Culture and Materials
4.2. Silencing
4.3. Western Blot Analysis
4.4. RTqPCR
4.5. Characterization of the Cellular Bioenergetics
4.6. Lactate Assay
4.7. Determination of Reactive Oxygen Species
4.8. Adenine Nucleotides Measurement
4.9. Cell Proliferation
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruprecht, J.J.; Kunji, E.R.S. Structural mechanism of transport of mitochondrial carriers. Annu. Rev. Biochem. 2021, 90, 535–558. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Locke, R.M. Thermogenic mechanisms in brown fat. Physiol. Rev. 1984, 64, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Bouillaud, F.; Couplan, E.; Pecqueur, C.; Ricquier, D. Homologues of the uncoupling protein from brown adipose tissue (UCP1): UCP2, UCP3, BMCP1 and UCP4. Biochim. Biophys. Acta 2001, 1504, 107–119. [Google Scholar] [CrossRef]
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Cannon, B.; Shabalina, I.G.; Kramarova, T.V.; Petrovic, N.; Nedergaard, J. Uncoupling proteins: A role in protection against reactive oxygen species—Or not? Biochim. Biophys. Acta 2006, 1757, 449–458. [Google Scholar] [CrossRef]
- Bouillaud, F. UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim. Biophys. Acta 2009, 1787, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, I.G.; Nedergaard, J. Mitochondrial (‘mild’) uncoupling and ROS production: Physiologically relevant or not? Biochem. Soc. Trans. 2011, 39, 1305–1309. [Google Scholar] [CrossRef]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef]
- Bouillaud, F.; Alves-Guerra, M.C.; Ricquier, D. UCPs, at the interface between bioenergetics and metabolism. Biochim. Biophys. Acta 2016, 1863, 2443–2456. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. Mitochondrial proton leaks and uncoupling proteins. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148428. [Google Scholar] [CrossRef] [PubMed]
- Hirschenson, J.; Melgar-Bermudez, E.; Mailloux, R.J. The uncoupling proteins: A systematic review on the mechanism used in the prevention of oxidative stress. Antioxidants 2022, 11, 322. [Google Scholar] [CrossRef] [PubMed]
- Luby, A.; Alves-Guerra, M.C. UCP2 as a cancer target through energy metabolism and oxidative stress control. Int. J. Mol. Sci. 2022, 23, 15077. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.E.; Antoniou, A.; Villalobos-Menuey, E.; Russo, A.; Trauger, R.; Vendemelio, M.; George, A.; Bartholomew, R.; Carlo, D.; Shaikh, A.; et al. Characterization of a novel metabolic strategy used by drug-resistant tumor cells. FASEB J. 2002, 16, 1550–1557. [Google Scholar] [CrossRef]
- Samudio, I.; Fiegl, M.; Andreeff, M. Mitochondrial uncoupling and the Warburg effect: Molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009, 69, 2163–2166. [Google Scholar] [CrossRef]
- Baffy, G. Uncoupling protein-2 and cancer. Mitochondrion 2021, 10, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G.; Derdak, Z.; Robson, S.C. Mitochondrial recoupling: A novel therapeutic strategy for cancer? Br. J. Cancer 2011, 105, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Esteves, P.; Pecqueur, C.; Ransy, C.; Esnous, C.; Lenoir, V.; Bouillaud, F.; Bulteau, A.L.; Lombès, A.; Prip-Buus, C.; Ricquier, D.; et al. Mitochondrial retrograde signaling mediated by UCP2 inhibits cancer cell proliferation and tumorigenesis. Cancer Res. 2014, 74, 3971–3982. [Google Scholar] [CrossRef]
- Sreedhar, A.; Petruska, P.; Miriyala, S.; Panchatcharam, M.; Zhao, Y. UCP2 overexpression enhanced glycolysis via activation of PFKFB2 during skin cell transformation. Oncotarget 2017, 8, 95504–95515. [Google Scholar] [CrossRef]
- Rupprecht, A.; Moldzio, R.; Mödl, B.; Pohl, E.E. Glutamine regulates mitochondrial uncoupling protein 2 to promote glutaminolysis in neuroblastoma cells. Biochim. Biophys. Acta 2019, 1860, 391–401. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, D.E. The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 1968, 7, 4030–4034. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.; George, L.K.; Grant, G.D.; Perou, C.M. Common markers of proliferation. Nat. Rev. Cancer 2006, 6, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Rial, E.; Rodríguez-Sánchez, L.; Aller, P.; Guisado, A.; González-Barroso, M.M.; Gallardo-Vara, E.; Redondo-Horcajo, M.; Castellanos, E.; Fernández de la Pradilla, R.; Viso, A. Development of chromanes as novel inhibitors of the uncoupling proteins. Chem. Biol. 2011, 18, 264–274. [Google Scholar] [CrossRef]
- Fleury, C.; Neverova, M.; Collins, S.; Raimbault, S.; Champigny, O.; Levi-Meyrueis, C.; Bouillaud, F.; Seldin, M.F.; Surwit, R.S.; Ricquier, D.; et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997, 15, 269–272. [Google Scholar] [CrossRef]
- Ricquier, D.; Bouillaud, F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem. J. 2000, 345, 161–179. [Google Scholar] [CrossRef]
- Zhou, Y.T.; Shimabukuro, M.; Koyama, K.; Lee, Y.; Wang, M.Y.; Trieu, F.; Newgard, C.B.; Unger, R.H. Induction by leptin of uncoupling protein-2 and enzymes of fatty acid oxidation. Proc. Natl. Acad. Sci. USA 1997, 94, 6386–6390. [Google Scholar] [CrossRef]
- Nègre-Salvayre, A.; Hirtz, C.; Carrera, G.; Cazenave, R.; Troly, M.; Salvayre, R.; Pénicaud, L.; Casteilla, L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 1997, 11, 809–815. [Google Scholar] [CrossRef]
- Anedda, A.; Rial, E.; González-Barroso, M.M. Metformin induces oxidative stress in white adipocytes and raises uncoupling protein 2 levels. J. Endocrinol. 2008, 199, 33–40. [Google Scholar] [CrossRef]
- Brand, M.D.; Esteves, T.C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005, 2, 85–93. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 1018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Cho, Y.S.; Jung, K.H.; Park, J.W.; Lee, K.H. Genipin enhances the antitumor effect of elesclomol in A549 lung cancer cells by blocking uncoupling protein-2 and stimulating reactive oxygen species production. Oncol. Lett. 2020, 20, 374. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys. 1996, 29, 169–202. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Brand, M.D. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem. Soc. Trans. 2003, 31, 1300–1301. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef]
- Nicholls, D.G. Mitochondrial membrane potential and aging. Aging Cell 2004, 3, 35–40. [Google Scholar] [CrossRef]
- Couplan, E.; Gonzalez-Barroso, M.M.; Alves-Guerra, M.C.; Ricquier, D.; Goubern, M.; Bouillaud, F. No evidence for a basal, retinoic, or superoxide-induced uncoupling activity of the uncoupling protein 2 present in spleen or lung mitochondria. J. Biol. Chem. 2002, 277, 26268–26275. [Google Scholar] [CrossRef]
- Kukat, A.; Dogan, S.A.; Edgar, D.; Mourier, A.; Jacoby, C.; Maiti, P.; Mauer, J.; Becker, C.; Senft, K.; Wibom, R.; et al. Loss of UCP2 attenuates mitochondrial dysfunction without altering ROS production and uncoupling activity. PLoS Genet. 2014, 10, e1004385. [Google Scholar] [CrossRef]
- Mattiasson, G.; Shamloo, M.; Gido, G.; Mathi, K.; Tomasevic, G.; Yi, S.; Warden, C.H.; Castilho, R.F.; Melcher, T.; Gonzalez-Zulueta, M.; et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med. 2003, 9, 1062–1068. [Google Scholar] [CrossRef]
- Carretero, M.V.; Torres, L.; Latasa, U.; García-Trevijano, E.R.; Prieto, J.; Mato, J.M.; Avila, M.A. Transformed but not normal hepatocytes express UCP2. FEBS Lett. 1998, 439, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Derdak, Z.; Mark, N.M.; Beldi, G.; Robson, S.C.; Wands, J.R.; Baffy, G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 2008, 68, 2813–2819. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. The influence of respiration and ATP hydrolysis on the proton electrochemical potential gradient across the inner membrane of rat liver mitochondria as determined by ion distribution. Eur. J. Biochem. 1974, 50, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. The effective proton conductances of the inner membrane of mitochondria from brown adipose tissue: Dependency on proton electrochemical gradient. Eur. J. Biochem. 1977, 77, 349–356. [Google Scholar] [CrossRef]
- Pons, P.G.; Nadal-Serrano, M.; Torrens-Mas, M.; Valle, A.; Oliver, J.; Roca, P. UCP2 inhibition sensitizes breast cancer cells to therapeutic agents by increasing oxidative stress. Free Radic. Biol. Med. 2015, 86, 67–77. [Google Scholar] [CrossRef]
- Brandi, J.; Cecconi, D.; Cordani, M.; Torrens-Mas, M.; Pacchiana, R.; Dalla Pozza, E.; Butera, G.; Manfredi, M.; Marengo, E.; Oliver, J.; et al. The antioxidant uncoupling protein 2 stimulates hnRNPA2/B1, GLUT1 and PKM2 expression and sensitizes pancreas cancer cells to glycolysis inhibition. Free Radic. Biol. Med. 2016, 101, 305–316. [Google Scholar] [CrossRef]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, A.; Piazzolla, C.; De Leonardis, F.; Gorgoglione, R.; Scarcia, P.; Pezzuto, F.; Agrimi, G.; et al. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat. Metab. 2020, 2, 1373–1381. [Google Scholar] [CrossRef]
- Hurtaud, C.; Gelly, C.; Chen, Z.; Lévi-Meyrueis, C.; Bouillaud, F. Glutamine stimulates translation of uncoupling protein 2 mRNA. Cell. Mol. Life Sci. 2007, 64, 1853–1860. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, Y.; Wu, S. The Warburg effect: Evolving interpretations of an established concept. Free Radic. Biol. Med. 2015, 79, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Boese, A.C.; Kang, S. Mitochondrial metabolism-mediated redox regulation in cancer progression. Redox Biol. 2021, 42, 101870. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Ali, H.; Du, Z.; Li, X.; Yang, Q.; Zhang, Y.C.; Wu, M.; Zhang, G. Identification of suitable reference genes for gene expression studies using quantitative polymerase chain reaction in lung cancer in vitro. Mol. Med. Rep. 2015, 11, 3767–3773. [Google Scholar] [CrossRef]
- De Korte, D.; Haverkort, W.A.; van Gennip, A.H.; Roos, D. Nucleotide profiles of normal human blood cells determined by high-performance liquid chromatography. Anal. Biochem. 1985, 147, 197–209. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segalés, J.; Sánchez-Martín, C.; Pujol-Morcillo, A.; Martín-Ruiz, M.; de los Santos, P.; Lobato-Alonso, D.; Oliver, E.; Rial, E. Role of UCP2 in the Energy Metabolism of the Cancer Cell Line A549. Int. J. Mol. Sci. 2023, 24, 8123. https://doi.org/10.3390/ijms24098123
Segalés J, Sánchez-Martín C, Pujol-Morcillo A, Martín-Ruiz M, de los Santos P, Lobato-Alonso D, Oliver E, Rial E. Role of UCP2 in the Energy Metabolism of the Cancer Cell Line A549. International Journal of Molecular Sciences. 2023; 24(9):8123. https://doi.org/10.3390/ijms24098123
Chicago/Turabian StyleSegalés, Jessica, Carlos Sánchez-Martín, Aleida Pujol-Morcillo, Marta Martín-Ruiz, Patricia de los Santos, Daniel Lobato-Alonso, Eduardo Oliver, and Eduardo Rial. 2023. "Role of UCP2 in the Energy Metabolism of the Cancer Cell Line A549" International Journal of Molecular Sciences 24, no. 9: 8123. https://doi.org/10.3390/ijms24098123
APA StyleSegalés, J., Sánchez-Martín, C., Pujol-Morcillo, A., Martín-Ruiz, M., de los Santos, P., Lobato-Alonso, D., Oliver, E., & Rial, E. (2023). Role of UCP2 in the Energy Metabolism of the Cancer Cell Line A549. International Journal of Molecular Sciences, 24(9), 8123. https://doi.org/10.3390/ijms24098123