
Cytokine Signatures in Psoriatic Arthritis Patients Indicate Different Phenotypic Traits Comparing Responders and Non-Responders of IL-17A and TNFα Inhibitors

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Baseline Characteristics
2.2. Cytokine Signatures at Baseline in PsA Patients Initiating TNFi
2.3. Immune Signatures at Baseline in Patients Initiating IL-17Ai
2.4. Biomarker Change after Four Months in TNFi DAPSA50 Responders and Non-Responders
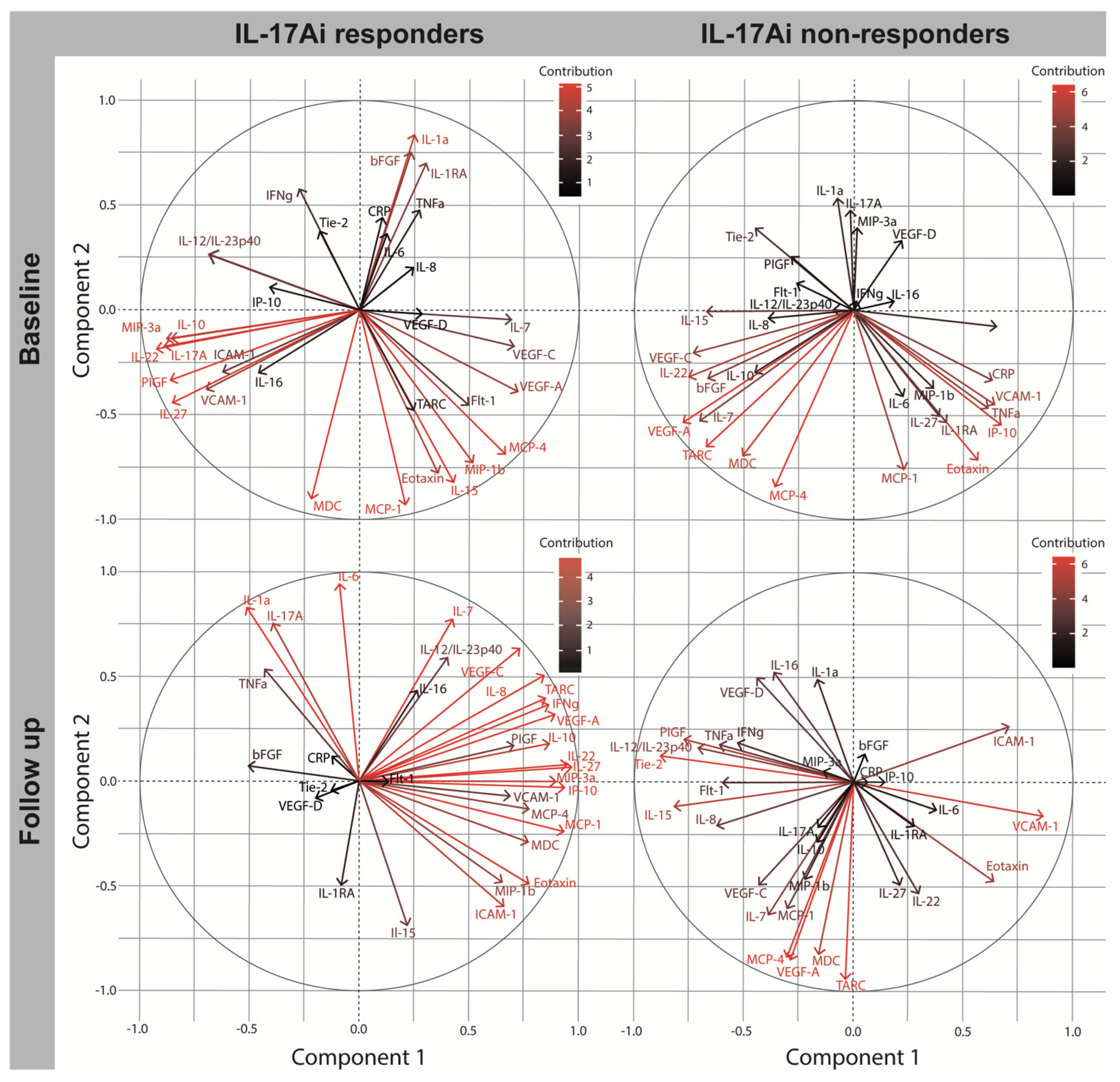
2.5. Biomarker Change after Four Months in IL17Ai DAPSA50 Responders and Non-Responders
2.6. Baseline Characteristics of MTX Initiators
3. Discussion
4. Materials and Methods
4.1. Study Setup and Participants
4.2. Clinical Examination and Preparation of Blood
4.3. Analysis of Plasma Markers
4.4. Statistical Analysis
4.5. Principal Component Analysis Correlation Plot
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGonagle, D. Enthesitis: An autoinflammatory lesion linking nail and joint involvement in psoriatic disease. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 9–13. [Google Scholar] [CrossRef]
- McGonagle, D.; McDermott, M.F. A proposed classification of the immunological diseases. PLoS Med. 2006, 3, 1242–1248. [Google Scholar] [CrossRef] [Green Version]
- Ritchlin, C.T.; Colbert, R.A.G.D.D. Psoriatic arthritis. N. Engl. J. Med. 2017, 376, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Kirkham, B.W.; Kavanaugh, A.; Reich, K. Interleukin-17A: A unique pathway in immune-mediated diseases: Psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology 2014, 141, 133–142. [Google Scholar] [CrossRef]
- Veale, D.J.; Fearon, U. The pathogenesis of psoriatic arthritis. Lancet 2018, 391, 2273–2284. [Google Scholar] [CrossRef]
- Aggarwal, A.; Agarwal, S.; Misra, R. Chemokine and chemokine receptor analysis reveals elevated interferon-inducible protein-10 (IP)-10/CXCL10 levels and increased number of CCR5+ and CXCR3+ CD4 T cells in synovial fluid of patients with enthesitis-related arthritis (ERA). Clin. Exp. Immunol. 2007, 148, 515–519. [Google Scholar] [CrossRef]
- Penkava, F.; Velasco-Herrera, M.D.C.; Young, M.D.; Yager, N.; Nwosu, L.N.; Pratt, A.G.; Lara, A.L.; Guzzo, C.; Maroof, A.; Mamanova, L.; et al. Single-cell sequencing reveals clonal expansions of pro-inflammatory synovial CD8 T cells expressing tissue-homing receptors in psoriatic arthritis. Nat. Commun. 2020, 11, 4767. [Google Scholar] [CrossRef] [PubMed]
- Wehr, P.; Purvis, H.; Law, S.; Thomas, R. Dendritic cells, T cells and their interaction in rheumatoid arthritis. Clin. Exp. Immunol. 2019, 196, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hueber, A.J.; McInnes, I.B. Immune regulation in psoriasis and psoriatic arthritis-Recent developments. Immunol. Lett. 2007, 114, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.; Liu, X.; Fang, L.; Chen, X.; Guo, T.; Zhang, J. The cytokine milieu in the interplay of pathogenic Th1/Th17 cells and regulatory T cells in autoimmune disease. Cell. Mol. Immunol. 2010, 7, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Silvagni, E.; Missiroli, S.; Perrone, M.; Patergnani, S.; Boncompagni, C.; Bortoluzzi, A.; Govoni, M.; Giorgi, C.; Alivernini, S.; Pinton, P.; et al. From Bed to Bench and Back: TNF-α, IL-23/IL-17A, and JAK-Dependent Inflammation in the Pathogenesis of Psoriatic Synovitis. Front. Pharmacol. 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chao, R.; Kavanaugh, A. Psoriatic Arthritis: Newer and Older Therapies. Curr. Rheumatol. Rep. 2019, 21, 75. [Google Scholar] [CrossRef]
- Kuek, A.; Hazleman, B.L.; Östör, A.J.K. Immune-mediated inflammatory diseases (IMIDs) and biologic therapy: A medical revolution. Postgrad. Med. J. 2007, 83, 251–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.L. Biologics-based therapy for the treatment of rheumatoid arthritis. Clin. Pharmacol. 2012, 91, 30–43. [Google Scholar] [CrossRef]
- Furue, K.; Ito, T.; Furue, M. Differential efficacy of biologic treatments targeting the TNF-α/IL-23/IL-17 axis in psoriasis and psoriatic arthritis. Cytokine 2018, 111, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.E.; Lie, E.; Jacobsson, L.T.; Christensen, R.; Mease, P.J.; Bliddal, H.; Geborek, P. Effectiveness and feasibility associated with switching to a second or third TNF inhibitor in patients with psoriatic arthritis: A cohort study from southern Sweden. J. Rheumatol. 2016, 43, 81–87. [Google Scholar] [CrossRef]
- Ko, J.M.; Gottlieb, A.B.; Kerbleski, J.F. Induction and exacerbation of psoriasis with TNF-blockade therapy: A review and analysis of 127 cases. J. Dermatol. Treat. 2009, 20, 100–108. [Google Scholar] [CrossRef]
- Meka, R.R.; Venkatesha, S.H.; Dudics, S.; Acharya, B.; Moudgil, K.D. IL-27-induced modulation of autoimmunity and its therapeutic potential. Autoimmun. Rev. 2015, 14, 1131–1141. [Google Scholar] [CrossRef] [Green Version]
- Muntyanu, A.; Abji, F.; Liang, K.; Pollock, R.A.; Chandran, V.; Gladman, D.D. Differential gene and protein expression of chemokines and cytokines in synovial fluid of patients with arthritis. Arthritis Res. 2016, 18, 296. [Google Scholar] [CrossRef] [Green Version]
- Diehl, S.; Rincón, M. The two faces of IL-6 on Th1/Th2 differentiation. Mol. Immunol. 2002, 39, 531–536. [Google Scholar] [CrossRef]
- Ogata, A.; Kumanogoh, A.; Tanaka, T. Pathological Role of Interleukin-6 in Psoriatic Arthritis. Arthritis 2012, 2012, 713618. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Hunter, C.A. The immunobiology of interleukin-27. Annu. Rev. Immunol. 2015, 33, 417–443. [Google Scholar] [CrossRef]
- van Roon, J.A.G.; Glaudemans, K.A.F.M.; Bijlsma, J.W.J.; Lafeber, F.P.J.G. Interleukin 7 stimulates tumour necrosis factor α and Th 1 cytokine production in joints of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Mavropoulos, A.; Varna, A.; Zafiriou, E.; Liaskos, C.; Alexiou, I.; Roussaki-Schulze, A.; Vlychou, M.; Katsiari, C.; Bogdanos, D.P.; Sakkas, L.I. IL-10 producing Bregs are impaired in psoriatic arthritis and psoriasis and inversely correlate with IL-17- and IFNγ-producing T cells. Clin. Immunol. 2017, 184, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Dayer, J.M.; Oliviero, F.; Punzi, L. A brief history of IL-1 and IL-1 Ra in rheumatology. Front. Pharmacol. 2017, 8, 293. [Google Scholar] [CrossRef] [Green Version]
- Vazirinejad, R.; Ahmadi, Z.; Kazemi Arababadi, M.; Hassanshahi, G.; Kennedy, D. The biological functions, structure and sources of CXCL10 and its outstanding part in the pathophysiology of multiple sclerosis. Neuroimmunomodulation 2014, 21, 322–330. [Google Scholar] [CrossRef]
- Aschard, H.; Vilhjálmsson, B.J.; Greliche, N.; Morange, P.-E.; Trégouët, D.-A.; Kraft, P. Maximizing the power of principal-component analysis of correlated phenotypes in genome-wide association studies. Am. J. Hum. Genet. 2014, 94, 662–676. [Google Scholar] [CrossRef] [Green Version]
- Ye, W.; Lu, W.; Tang, Y.; Chen, G.; Li, X.; Ji, C.; Hou, M.; Zeng, G.; Lan, X.; Wang, Y.; et al. Identification of COVID-19 Clinical Phenotypes by Principal Component Analysis-Based Cluster Analysis. Front. Med. 2020, 7, 570614. [Google Scholar] [CrossRef] [PubMed]
- Merola, J.F.; Espinoza, L.R.; Fleischmann, R. Distinguishing rheumatoid arthritis from psoriatic arthritis. RMD Open 2018, 4, e000656. [Google Scholar] [CrossRef]
- Elkassar, N.; Gress, R.E. An overview of IL-7 biology and its use in immunotherapy. J. Immunotoxicol. 2010, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, R.S.G.; Duarte, A.L.B.P. IL-7 is a key driver cytokine in spondyloarthritis? J. Immunol. Res. 2019, 2019, 7453236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benham, H.; Norris, P.; Goodall, J.; Wechalekar, M.D.; FitzGerald, O.; Szentpetery, A.; Smith, M.; Thomas, R.; Gaston, H. Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res. Ther. 2013, 15, R136. [Google Scholar] [CrossRef] [Green Version]
- Iannis, E.A.; Pflanz, S. The emerging role of Interleukin 27 in inflammatory arthritis and bone destruction. Cytokine Growth Factor Rev. 2013, 24, 115–121. [Google Scholar] [CrossRef]
- Qin, H.; Wang, L.; Feng, T.; Elson, C.O.; Niyongere, S.A.; Lee, S.J.; Reynolds, S.L.; Weaver, C.T.; Roarty, K.; Serra, R.; et al. TGF-β Promotes Th17 Cell Development through Inhibition of SOCS3. J. Immunol. 2009, 183, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Blauvelt, A.; Chiricozzi, A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clin. Rev. Allergy Immunol. 2018, 55, 379–390. [Google Scholar] [CrossRef] [Green Version]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.M.; McClanahan, T.K.; Cua, D.J. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Maria Patron, R.R.D.D.S. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 71, 5–17. [Google Scholar] [CrossRef]
- Lee, H.L.; Jang, J.W.; Lee, S.W.; Yoo, S.H.; Kwon, J.H.; Nam, S.W.; Bae, S.H.; Choi, J.Y.; Han, N.I.; Yoon, S.K. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci. Rep. 2019, 9, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.-C.; Young, H.A. The talented interferon-gamma. Adv. Biosci. Biotechnol. 2013, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Yeh, W.-I.; McWilliams, I.L.; Harrington, L.E. IFNγ inhibits Th17 differentiation and function via Tbet-dependent and Tbet-independent mechanisms. J. Neuroimmunol. 2014, 267, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Skougaard, M.; Ditlev, S.B.; Stisen, Z.R.; Coates, L.C.; Ellegaard, K.; Kristensen, L.E. Four emerging immune cellular blood phenotypes associated with disease duration and activity established in Psoriatic Arthritis. Arthritis Res. Ther. 2022, 24, 262. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, S.; Gehrmann, U.; Preite, S.; Mitra, S. Cytokine-regulated Th17 plasticity in human health and diseases. Immunology 2021, 163, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Nistala, K.; Adams, S.; Cambrook, H.; Ursu, S.; Olivito, B.; de Jager, W.; Evans, J.G.; Cimaz, R.; Bajaj-Elliott, M.; Wedderburn, L.R. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl. Acad. Sci. USA 2010, 107, 14751–14756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, H.M.; Pan, N.; Lehman, T.J.; Adams, A.; Kalliolias, G.D.; Zhu, Y.; Santiago, F.; Nguyen, J.; Sitaras, L.; Cunningham-Rundles, S.; et al. The impact of disease activity and tumour necrosis factor-α inhibitor therapy on cytokine levels in juvenile idiopathic arthritis. Clin. Exp. Immunol. 2016, 184, 308–317. [Google Scholar] [CrossRef] [Green Version]
- Schulz, M.; Dotzlaw, H.; Neeck, G. Ankylosing spondylitis and rheumatoid arthritis: Serum levels of TNF- α and its soluble receptors during the course of therapy with etanercept and infliximab. Biomed. Res. Int. 2014, 2014, 675108. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Rudwaleit, M.; Brandt, J.; Thiel, A.; Braun, J.; Sieper, J. Up regulation of the production of tumour necrosis factor α and interferon γ by T cells in ankylosing spondylitis during treatment with etanercept. Ann. Rheum. Dis. 2003, 62, 561–564. [Google Scholar] [CrossRef] [Green Version]
- Schoels, M.M.; Aletaha, D.; Alasti, F.; Smolen, J.S. Disease activity in psoriatic arthritis (PsA): Defining remission and treatment success using the DAPSA score. Ann. Rheum. Dis. 2016, 75, 811–818. [Google Scholar] [CrossRef]
- Gossec, L.; Smolen, J.S.; Ramiro, S.; De Wit, M.; Cutolo, M.; Dougados, M.; Emery, P.; Landewé, R.; Oliver, S.; Aletaha, D.; et al. European League Against Rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Ann. Rheum. Dis. 2016, 75, 499–510. [Google Scholar] [CrossRef]
- Højgaard, P.; Christensen, R.; Dreyer, L.; Mease, P.; de Wit, M.; Skov, L.; Glintborg, B.; Christensen, A.W.; Ballegaard, C.; Bliddal, H.; et al. Pain mechanisms and ultrasonic inflammatory activity as prognostic factors in patients with psoriatic arthritis: Protocol for a prospective, exploratory cohort study. BMJ Open 2016, 6, e010650. [Google Scholar] [CrossRef] [Green Version]
- Nistor, G.I.; Dillman, R.O. Cytokine network analysis of immune responses before and after autologous dendritic cell and tumor cell vaccine immunotherapies in a randomized trial. J. Transl. Med. 2020, 18, 176. [Google Scholar] [CrossRef] [Green Version]
- Jolliffe, I.T. Rotation of principal components: Choice of normalization constraints. J. Appl. Stat. 1995, 22, 29–35. [Google Scholar] [CrossRef]
- Winchester, R.; FitzGerald, O. The many faces of psoriatic arthritis: Their genetic determinism. Rheumatology 2020, 59, i4–i9. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| TNFi Responders n = 11 | TNFi Non-Responders n = 9 | p-Value | IL-17i Responders n = 7 | IL-17i Non-Responders n = 12 | p-Value | MTX n = 10 | p-Value | |
|---|---|---|---|---|---|---|---|---|
| Female | 7 (63.6) | 5 (55.6) | 0.714 | 4 (57.1) | 8 (66.7) | 0.678 | 5 (50.0) | 0.787 † |
| Age | 48.9 (46.5–55.2) | 50.4 (45.5–61.4) | 0.470 | 48.4 (40.7–63.0) | 53.5 (45.2–60.8) | 0.939 | 61.0 (54.2–66.0) | 0.163 † |
| Disease duration | 4.4 (2.6–7.7) | 3.3 (2.1–17.0) | 0.970 | 9.8 (0.9–14.7) | 5.9 (2.3–13.7) | 0.929 | 2.3 (1.1–10.6) | 0.692 † |
| Previous bDMARD | ||||||||
| 0 | 8 (72.7) | 5 (55.6) | 0.437 | 2 (28.6) | 4 (33.3) | 0.792 | 9 (90.0) | 0.049 † |
| 1 | 3 (27.3) | 2 (22.2) | 1 (14.3) | 4 (33.3) | 1 (10.0) | |||
| 2 | 0 (0.0) | 1 (11.1) | 2 (28.6) | 2 (16.7) | 0 (0.0) | |||
| ≥3 | 0 (0.0) | 1 (11.1) | 2 (28.6) | 2 (16.7) | 0 (0.0) | |||
| Concomitant csDMARD | ||||||||
| 0 | 9 (0.8) | 4 (0.45) | 0.134 | 4 (0.6) | 6 (0.5) | 0.882 | 0 (0.0) | <0.001 † |
| MTX | 1 (0.1) | 4 (0.45) | 2 (0.3) | 3 (0.3) | 0 (0.0) | |||
| SSZ | 1 (0.1) | 0 (0.0) | 0 (0.0) | 1 (0.1) | 0 (0.0) | |||
| LEF | 0 (0.0) | 1 (0.1) | 1 (0.1) | 2 (0.2) | 0 (0.0) | |||
| DAPSA | 27.0 (22.6–41.3) | 38.5 (22.9–39.5) | 0.676 | 37.2 (24.2–38.7) | 29.5 (26.2–47.3) | 0.708 | 25.0 (18.1–34.8) | 0.208 † |
| SJC (66) | 5.0 (4.5–10.5) | 7.0 (3.0–8.0) | 0.321 | 5.0 (4.5–5.5) | 6.0 (3.0–11.3) | 0.977 | 6.0 (3.8–8.5) | 0.993 † |
| TJC (68) | 7.0 (5.0–12.5) | 19.0 (10.0–29.0) | 0.303 | 12.0 (6.0–19.0) | 10.0 (8.3–24.0) | 0.547 | 7.5 (5.5–11.0) | 0.286 † |
| SPARCC | 3.0 (1.0–6.0) | 5.0 (4.0–6.0) | 0.514 | 4.0 (1.5–8.0) | 3.0 (1.8–5.0) | 0.435 | 2.5 (0.3–3.0) | 0.204 † |
| US score | 8.0 (2.5–8.5) | 6.0 (4.0–8.0) | 0.508 | 5.0 (4.5–5.5) | 4.5 (1.0–6.3) | 0.108 | 8.0 (5.0–9.0) | 0.137 † |
| PASI | 1.5 (0.2–2.5) | 1.5 (1.2–5.0) | 0.401 | 1.4 (0.0–2.0) | 0.4 (0.0–2.2) | 0.930 | 1.0 (0.7–3.2) | 0.416 † |
| Baseline | Follow Up | |||||||
|---|---|---|---|---|---|---|---|---|
| TNFi Responders | TNFi Non-Responders | TNFi Responders | TNFi Non-Responders | |||||
| Biomarker | C1 | C2 | C1 | C2 | C1 | C2 | C1 | C2 |
| bFGF | −0.382 | 0.465 | −0.308 | 0.100 | 0.552 | 0.730 | 0.574 | −0.644 |
| Flt-1 | −0.424 | 0.288 | 0.757 | 0.499 | −0.087 | 0.651 | 0.464 | 0.393 |
| PlGF | −0.348 | 0.168 | 0.382 | 0.690 | 0.554 | 0.365 | 0.410 | 0.579 |
| Tie-2 | −0.532 | 0.044 | 0.378 | 0.642 | −0.177 | 0.438 | 0.055 | 0.296 |
| VEGF-A | 0.010 | 0.516 | 0.796 | −0.171 | 0.911 | −0.065 | 0.589 | −0.482 |
| VEGF-C | −0.469 | 0.549 | 0.883 | 0.066 | 0.192 | 0.289 | 0.662 | 0.124 |
| VEGF-D | 0.075 | 0.012 | 0.259 | −0.049 | 0.403 | −0.196 | −0.126 | 0.694 |
| Eotaxin | 0.869 | 0.002 | 0.840 | −0.338 | 0.524 | −0.526 | 0.919 | 0.054 |
| IP-10 | 0.867 | 0.436 | 0.426 | 0.424 | 0.360 | −0.224 | 0.316 | 0.614 |
| MCP-1 | 0.842 | −0.272 | 0.934 | −0.310 | 0.841 | −0.387 | 0.722 | −0.152 |
| MCP-4 | 0.608 | −0.039 | 0.806 | −0.182 | 0.933 | −0.121 | 0.756 | −0.563 |
| MDC | 0.579 | −0.508 | 0.314 | −0.373 | 0.906 | −0.142 | 0.728 | −0.545 |
| MIP-1b | 0.912 | 0.191 | 0.858 | −0.392 | 0.754 | −0.508 | 0.601 | 0.231 |
| TARC | 0.769 | 0.061 | 0.473 | 0.177 | 0.878 | −0.073 | 0.709 | −0.638 |
| IL12/IL23p40 | 0.020 | 0.663 | −0.446 | 0.273 | 0.059 | −0.708 | 0.127 | 0.597 |
| IL-15 | 0.166 | −0.036 | 0.533 | 0.540 | 0.728 | 0.441 | 0.706 | 0.185 |
| IL-16 | −0.212 | 0.592 | −0.011 | 0.806 | −0.227 | −0.309 | −0.073 | 0.750 |
| IL-1α | 0.157 | 0.162 | −0.402 | 0.039 | −0.543 | −0.703 | −0.761 | −0.135 |
| IL-7 | 0.108 | 0.609 | 0.908 | 0.013 | 0.897 | 0.205 | 0.817 | −0.309 |
| IL-17A | −0.029 | 0.455 | −0.090 | −0.184 | 0.145 | 0.347 | −0.839 | −0.442 |
| IL-1RA | 0.360 | −0.455 | 0.643 | 0.384 | 0.797 | −0.138 | −0.006 | −0.106 |
| IFNg | 0.761 | 0.531 | 0.736 | 0.181 | 0.704 | −0.399 | 0.319 | 0.758 |
| IL-10 | −0.437 | 0.039 | 0.635 | 0.122 | −0.542 | −0.116 | 0.647 | 0.280 |
| IL-6 | 0.712 | 0.559 | 0.044 | 0.879 | 0.610 | −0.325 | 0.442 | 0.309 |
| IL-8 | −0.124 | 0.846 | 0.743 | 0.535 | 0.388 | 0.296 | 0.409 | 0.387 |
| TNFa | −0.361 | 0.555 | 0.880 | −0.274 | 0.651 | 0.013 | 0.570 | 0.387 |
| IL-22 | 0.414 | −0.255 | 0.797 | −0.281 | 0.436 | 0.436 | 0.718 | −0.630 |
| IL-27 | 0.783 | −0.239 | 0.860 | −0.294 | 0.662 | −0.104 | 0.581 | 0.338 |
| MIP-3a | 0.248 | 0.474 | −0.450 | −0.052 | 0.445 | 0.429 | −0.675 | −0.114 |
| CRP | −0.515 | 0.456 | −0.147 | 0.210 | 0.783 | 0.190 | 0.163 | 0.296 |
| ICAM-1 | 0.791 | 0.497 | 0.432 | −0.266 | 0.655 | −0.230 | 0.121 | −0.050 |
| VCAM-1 | 0.770 | 0.413 | 0.908 | −0.083 | −0.134 | −0.795 | 0.411 | 0.123 |
| Baseline | Follow Up | |||||||
|---|---|---|---|---|---|---|---|---|
| IL-17Ai Responders | IL-17Ai Non-Responders | IL-17Ai Responders | IL-17Ai Non-Responders | |||||
| Biomarker | C1 | C2 | C1 | C2 | C1 | C2 | C1 | C2 |
| bFGF | 0.232 | 0.755 | −0.670 | −0.331 | −0.513 | 0.071 | 0.047 | 0.137 |
| Flt-1 | 0.501 | −0.459 | −0.263 | 0.125 | 0.139 | −0.004 | −0.602 | 0.000 |
| PlGF | −0.867 | −0.338 | −0.290 | 0.254 | 0.702 | 0.170 | −0.778 | 0.209 |
| Tie-2 | −0.189 | 0.381 | −0.453 | 0.391 | −0.134 | −0.048 | −0.886 | 0.129 |
| VEGF-A | 0.724 | −0.396 | −0.782 | −0.539 | 0.890 | 0.318 | −0.292 | −0.846 |
| VEGF-C | 0.708 | −0.178 | −0.736 | −0.206 | 0.729 | 0.634 | −0.435 | −0.488 |
| VEGF-D | 0.288 | −0.026 | 0.216 | 0.331 | −0.205 | −0.081 | −0.447 | 0.503 |
| Eotaxin | 0.349 | −0.779 | 0.564 | −0.721 | 0.770 | −0.493 | 0.636 | −0.472 |
| IP-10 | −0.415 | 0.107 | 0.671 | −0.552 | 0.935 | −0.032 | 0.143 | 0.003 |
| MCP-1 | 0.205 | −0.933 | 0.228 | −0.763 | 0.932 | −0.241 | −0.307 | −0.599 |
| MCP-4 | 0.665 | −0.692 | −0.361 | −0.847 | 0.771 | −0.136 | −0.308 | −0.836 |
| MDC | −0.224 | −0.907 | −0.509 | −0.670 | 0.769 | −0.292 | −0.162 | −0.821 |
| MIP-1β | 0.516 | −0.734 | 0.361 | −0.377 | 0.648 | −0.483 | −0.224 | −0.461 |
| TARC | 0.248 | −0.486 | −0.676 | −0.657 | 0.844 | 0.396 | −0.042 | −0.940 |
| IL12/IL23p40 | −0.693 | 0.263 | −0.096 | 0.004 | 0.400 | 0.589 | −0.712 | 0.167 |
| IL-15 | 0.424 | −0.827 | −0.676 | −0.010 | 0.213 | −0.691 | −0.824 | −0.114 |
| IL-16 | −0.464 | −0.304 | 0.183 | 0.040 | 0.262 | 0.433 | −0.369 | 0.528 |
| IL-1α | 0.249 | 0.840 | −0.077 | 0.534 | −0.518 | 0.829 | −0.169 | 0.493 |
| IL-7 | 0.696 | −0.050 | −0.711 | −0.532 | 0.423 | 0.772 | −0.393 | −0.631 |
| IL-17A | −0.894 | −0.177 | −0.018 | 0.479 | −0.399 | 0.755 | −0.165 | −0.214 |
| IL-1RA | 0.302 | 0.700 | 0.423 | −0.533 | −0.089 | −0.502 | 0.273 | −0.209 |
| IFNγ | −0.280 | 0.578 | 0.008 | 0.041 | 0.861 | 0.363 | −0.534 | 0.193 |
| IL-10 | −0.863 | −0.137 | −0.453 | −0.298 | 0.864 | 0.179 | −0.172 | −0.287 |
| IL-6 | 0.121 | 0.364 | 0.224 | −0.414 | −0.095 | 0.942 | 0.375 | −0.133 |
| IL-8 | 0.245 | 0.120 | −0.393 | −0.040 | 0.841 | 0.505 | −0.632 | −0.204 |
| TNFα | 0.275 | 0.476 | 0.609 | −0.470 | −0.436 | 0.535 | −0.616 | 0.181 |
| IL-22 | −0.930 | −0.188 | −0.757 | −0.322 | 0.951 | 0.079 | 0.292 | −0.530 |
| IL-27 | −0.857 | −0.448 | 0.391 | −0.513 | 0.967 | 0.065 | 0.208 | −0.490 |
| MIP-3α | −0.884 | −0.137 | 0.012 | 0.396 | 0.903 | −0.002 | −0.144 | 0.052 |
| CRP | 0.103 | 0.442 | 0.630 | −0.337 | −0.130 | 0.117 | 0.068 | 0.000 |
| ICAM-1 | −0.627 | −0.220 | 0.664 | −0.053 | 0.653 | −0.599 | 0.707 | 0.266 |
| VCAM-1 | −0.703 | −0.385 | 0.641 | −0.456 | 0.687 | −0.071 | 0.863 | −0.161 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skougaard, M.; Ditlev, S.B.; Søndergaard, M.F.; Kristensen, L.E. Cytokine Signatures in Psoriatic Arthritis Patients Indicate Different Phenotypic Traits Comparing Responders and Non-Responders of IL-17A and TNFα Inhibitors. Int. J. Mol. Sci. 2023, 24, 6343. https://doi.org/10.3390/ijms24076343
Skougaard M, Ditlev SB, Søndergaard MF, Kristensen LE. Cytokine Signatures in Psoriatic Arthritis Patients Indicate Different Phenotypic Traits Comparing Responders and Non-Responders of IL-17A and TNFα Inhibitors. International Journal of Molecular Sciences. 2023; 24(7):6343. https://doi.org/10.3390/ijms24076343
Chicago/Turabian StyleSkougaard, Marie, Sisse Bolm Ditlev, Magnus Friis Søndergaard, and Lars Erik Kristensen. 2023. "Cytokine Signatures in Psoriatic Arthritis Patients Indicate Different Phenotypic Traits Comparing Responders and Non-Responders of IL-17A and TNFα Inhibitors" International Journal of Molecular Sciences 24, no. 7: 6343. https://doi.org/10.3390/ijms24076343
APA StyleSkougaard, M., Ditlev, S. B., Søndergaard, M. F., & Kristensen, L. E. (2023). Cytokine Signatures in Psoriatic Arthritis Patients Indicate Different Phenotypic Traits Comparing Responders and Non-Responders of IL-17A and TNFα Inhibitors. International Journal of Molecular Sciences, 24(7), 6343. https://doi.org/10.3390/ijms24076343