

Neutrophils in Health and Disease: From Receptor Sensing to Inflammasome Activation

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Mechanisms of Recognition and Killing of Pathogens by Neutrophils
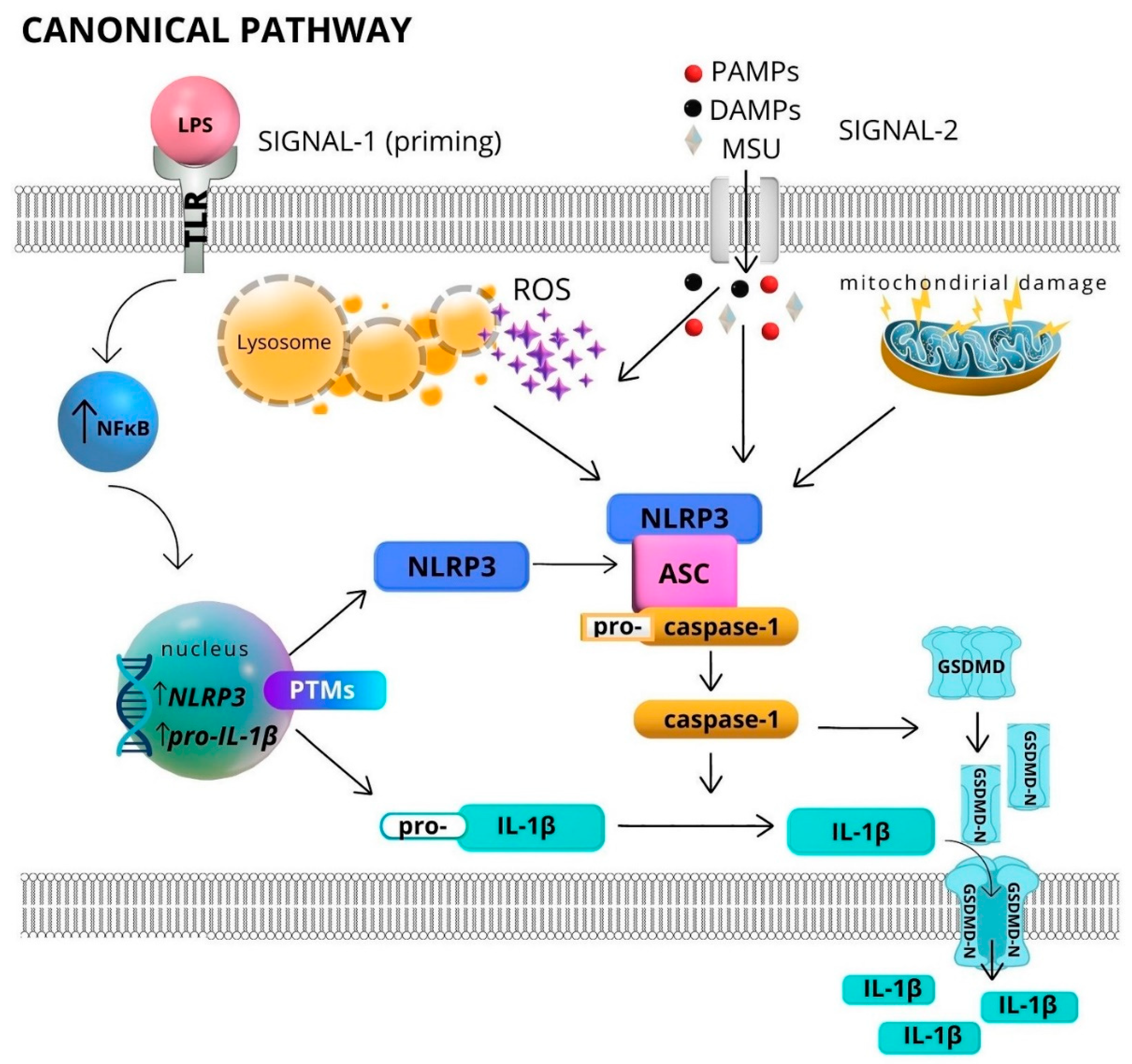
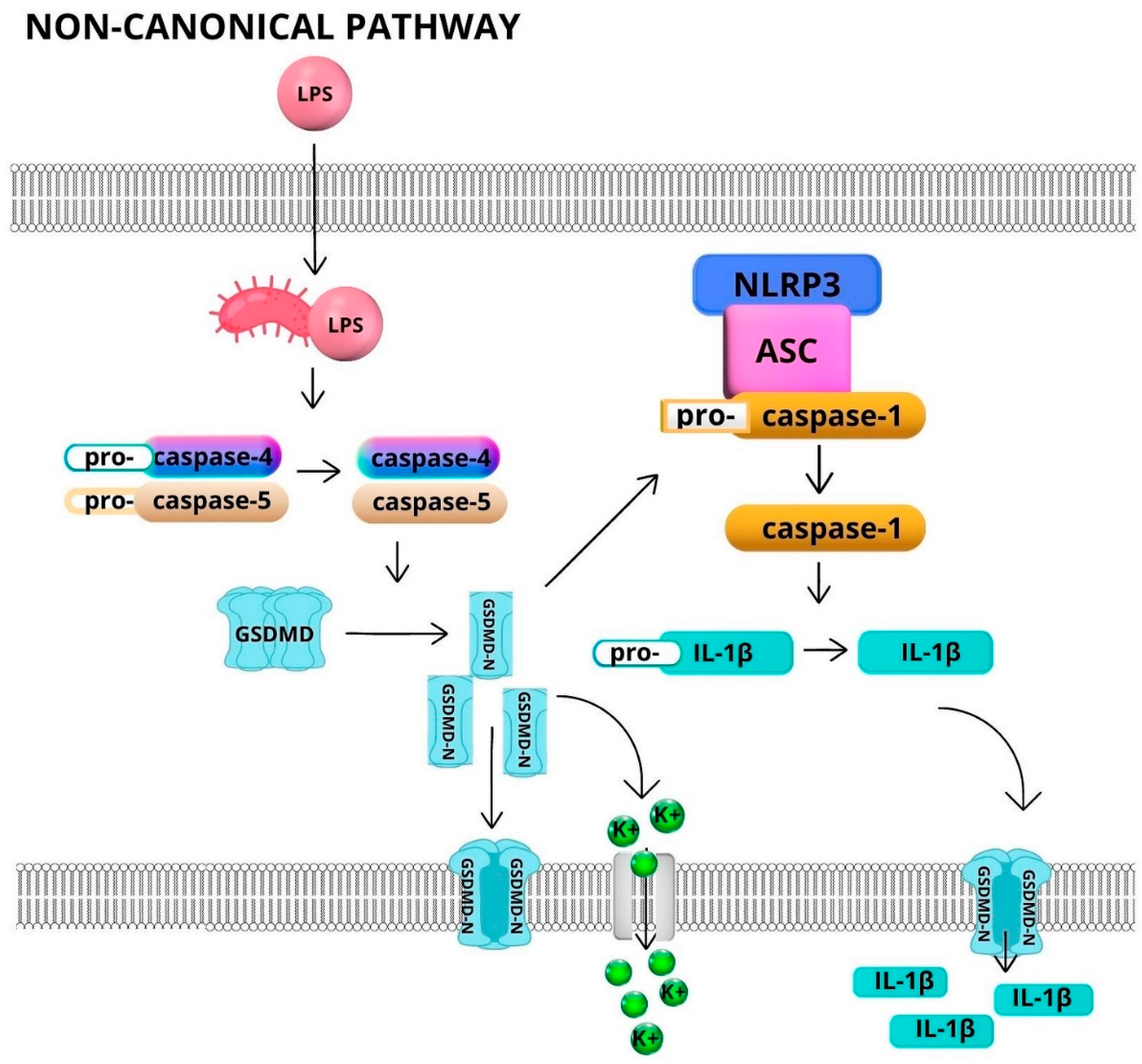
2. Inflammasomes—Structure and Activation
Activation of Inflammasomes in Neutrophils
- The presence of numerous spots of enzymatically active caspase-1 (confirmed with a FLICA test) in the PMNs of the infected cornea; and
- ASC staining in bone-marrow-derived neutrophils stimulated with thermally inactivated Streptococcus pneumoniae and pneumolysin (confirmed by positive FLICA test and fluorescence), indicating the presence of NLRP3.
3. Expression of IL-1β by Neutrophils with the Contribution of Inflammasomes
3.1. Inflammasome Activation with Canonical Pathway and Serine Proteases
3.2. Neutrophils vs. Pyroptosis—Nonlytic Expression of IL-1β
3.3. Non-canonical Activation of Inflammasome in Neutrophils—IL-1β and NETs
3.4. IL-1β Expression with Contribution of Autophagosomes
4. Inflammasomes in Neutrophils and Their Contribution to Disease Pathogenesis
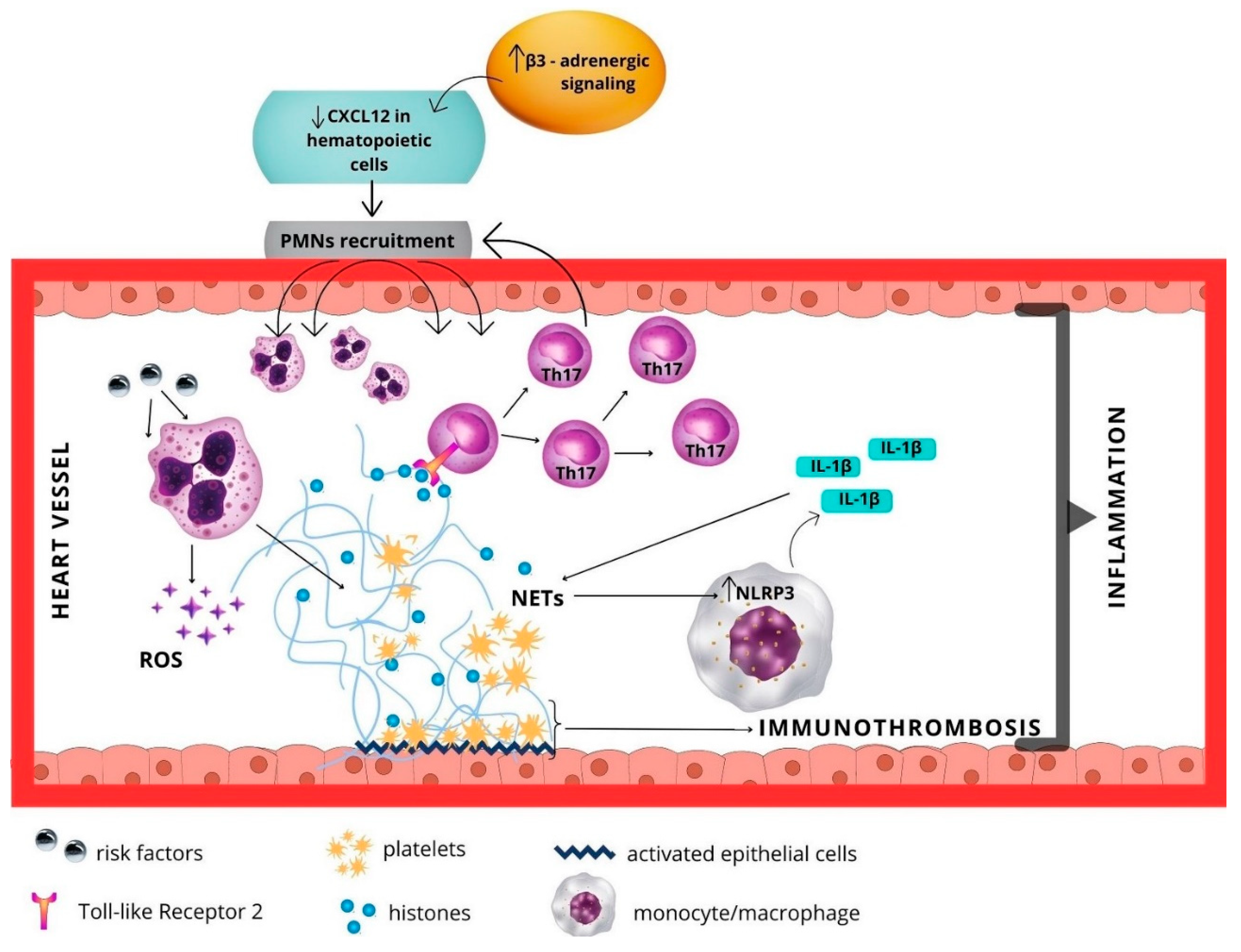
4.1. Cardiovascular Diseases
4.2. Severe Course of COVID-19
4.3. Cryopyrin-Associated Periodic Syndromes
5. Summary and Future Prospects
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mócsai, A. Diverse Novel Functions of Neutrophils in Immunity, Inflammation, and Beyond. J. Exp. Med. 2013, 210, 1283–1299. [Google Scholar] [CrossRef]
- Hayashi, F.; Means, T.K.; Luster, A.D. Toll-like Receptors Stimulate Human Neutrophil Function. Blood 2003, 102, 2660–2669. [Google Scholar] [CrossRef] [PubMed]
- El Kebir, D.; József, L.; Filep, J.G. Neutrophil Recognition of Bacterial DNA and Toll-like Receptor 9-Dependent and -Independent Regulation of Neutrophil Function. Arch. Immunol. Ther. Exp. (Warsz.) 2008, 56, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D.; Morgan, A. Secretory Granule Exocytosis. Physiol. Rev. 2003, 83, 581–632. [Google Scholar] [CrossRef] [PubMed]
- Lacy, P.; Eitzen, G. Control of Granule Exocytosis in Neutrophils. Front. Biosci. J. Virtual Libr. 2008, 13, 5559–5570. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.M. Phagocytic Leukocytes and Reactive Oxygen Species. Histochem. Cell Biol. 2009, 131, 465–469. [Google Scholar] [CrossRef]
- Almyroudis, N.G.; Grimm, M.J.; Davidson, B.A.; Röhm, M.; Urban, C.F.; Segal, B.H. NETosis and NADPH Oxidase: At the Intersection of Host Defense, Inflammation, and Injury. Front. Immunol. 2013, 4, 45. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil Extracellular Traps: Double-Edged Swords of Innate Immunity. J. Immunol. Baltim. Md 1950 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- Bakele, M.; Joos, M.; Burdi, S.; Allgaier, N.; Pöschel, S.; Fehrenbacher, B.; Schaller, M.; Marcos, V.; Kümmerle-Deschner, J.; Rieber, N.; et al. Localization and Functionality of the Inflammasome in Neutrophils. J. Biol. Chem. 2014, 289, 5320–5329. [Google Scholar] [CrossRef]
- Colotta, F.; Re, F.; Polentarutti, N.; Sozzani, S.; Mantovani, A. Modulation of Granulocyte Survival and Programmed Cell Death by Cytokines and Bacterial Products. Blood 1992, 80, 2012–2020. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of ProIL-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Nathan, C. Neutrophils and Immunity: Challenges and Opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Schroder, K. Antimicrobial Functions of Inflammasomes. Curr. Opin. Microbiol. 2013, 16, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF-ΚB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Greaney, A.J.; Leppla, S.H.; Moayeri, M. Bacterial Exotoxins and the Inflammasome. Front. Immunol. 2015, 6, 570. [Google Scholar] [CrossRef]
- Kanneganti, T.-D.; Ozören, N.; Body-Malapel, M.; Amer, A.; Park, J.-H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and Small Antiviral Compounds Activate Caspase-1 through Cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome through Phagosomal Destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Sborgi, L.; Rühl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Müller, D.J.; Broz, P.; Hiller, S. GSDMD Membrane Pore Formation Constitutes the Mechanism of Pyroptotic Cell Death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Song, N.; Liu, Z.-S.; Xue, W.; Bai, Z.-F.; Wang, Q.-Y.; Dai, J.; Liu, X.; Huang, Y.-J.; Cai, H.; Zhan, X.-Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197.e6. [Google Scholar] [CrossRef]
- Stutz, A.; Kolbe, C.-C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; van Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E. NLRP3 Inflammasome Assembly Is Regulated by Phosphorylation of the Pyrin Domain. J. Exp. Med. 2017, 214, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Castejon, G. Control of the Inflammasome by the Ubiquitin System. FEBS J. 2020, 287, 11–26. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Schmid-Burgk, J.L.; Gaidt, M.M.; Schmidt, T.; Ebert, T.S.; Bartok, E.; Hornung, V. Caspase-4 Mediates Non-Canonical Activation of the NLRP3 Inflammasome in Human Myeloid Cells. Eur. J. Immunol. 2015, 45, 2911–2917. [Google Scholar] [CrossRef]
- Chen, K.W.; Groß, C.J.; Sotomayor, F.V.; Stacey, K.J.; Tschopp, J.; Sweet, M.J.; Schroder, K. The Neutrophil NLRC4 Inflammasome Selectively Promotes IL-1β Maturation without Pyroptosis during Acute Salmonella Challenge. Cell Rep. 2014, 8, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sun, B. Neutrophil Pyroptosis: New Perspectives on Sepsis. Cell. Mol. Life Sci. 2019, 76, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential Expression of NLRP3 among Hematopoietic Cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef]
- Karmakar, M.; Katsnelson, M.; Malak, H.A.; Greene, N.G.; Howell, S.J.; Hise, A.G.; Camilli, A.; Kadioglu, A.; Dubyak, G.R.; Pearlman, E. Neutrophil IL-1β Processing Induced by Pneumolysin Is Mediated by the NLRP3/ASC Inflammasome and Caspase-1 Activation and Is Dependent on K+ Efflux. J. Immunol. Baltim. Md 1950 2015, 194, 1763–1775. [Google Scholar] [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 Self-Cleavage Is an Intrinsic Mechanism to Terminate Inflammasome Activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Wengner, A.M.; Pitchford, S.C.; Furze, R.C.; Rankin, S.M. The Coordinated Action of G-CSF and ELR + CXC Chemokines in Neutrophil Mobilization during Acute Inflammation. Blood 2008, 111, 42–49. [Google Scholar] [CrossRef]
- Kennedy, A.D.; DeLeo, F.R. Neutrophil Apoptosis and the Resolution of Infection. Immunol. Res. 2009, 43, 25–61. [Google Scholar] [CrossRef]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The Role of Interleukin-1 in General Pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [PubMed]
- Tourneur, L.; Witko-Sarsat, V. Inflammasome Activation: Neutrophils Go Their Own Way. J. Leukoc. Biol. 2019, 105, 433–436. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.-C.; Goode, J.; Miething, C.; Göktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-ΚB Is a Negative Regulator of IL-1β Secretion as Revealed by Genetic and Pharmacological Inhibition of IKKβ. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef]
- Mankan, A.K.; Dau, T.; Jenne, D.; Hornung, V. The NLRP3/ASC/Caspase-1 Axis Regulates IL-1β Processing in Neutrophils. Eur. J. Immunol. 2012, 42, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Keitelman, I.A.; Shiromizu, C.M.; Zgajnar, N.R.; Danielián, S.; Jancic, C.C.; Martí, M.A.; Fuentes, F.; Yancoski, J.; Vera Aguilar, D.; Rosso, D.A.; et al. The Interplay between Serine Proteases and Caspase-1 Regulates the Autophagy-Mediated Secretion of Interleukin-1 Beta in Human Neutrophils. Front. Immunol. 2022, 13, 832306. [Google Scholar] [CrossRef]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical Inflammasome Signaling Elicits Gasdermin D-Dependent Neutrophil Extracellular Traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D Plays a Vital Role in the Generation of Neutrophil Extracellular Traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 in the Pathogenesis and Treatment of Inflammatory Diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, M.; Stow, J.L.; Schroder, K. Mechanisms of Unconventional Secretion of IL-1 Family Cytokines. Cytokine 2015, 74, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraj, S.L.; Feltham, R.; Rashidi, M.; Frank, D.; Liu, Z.; Simpson, D.S.; Ebert, G.; Vince, A.; Herold, M.J.; Kueh, A.; et al. The Ubiquitylation of IL-1β Limits Its Cleavage by Caspase-1 and Targets It for Proteasomal Degradation. Nat. Commun. 2021, 12, 2713. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrín, P.; Shao, F. The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Yow, S.J.; Yeap, H.W.; Chen, K.W. Inflammasome and Gasdermin Signaling in Neutrophils. Mol. Microbiol. 2022, 117, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, M.; Minns, M.; Greenberg, E.N.; Diaz-Aponte, J.; Pestonjamasp, K.; Johnson, J.L.; Rathkey, J.K.; Abbott, D.W.; Wang, K.; Shao, F.; et al. N-GSDMD Trafficking to Neutrophil Organelles Facilitates IL-1β Release Independently of Plasma Membrane Pores and Pyroptosis. Nat. Commun. 2020, 11, 2212. [Google Scholar] [CrossRef] [PubMed]
- Iula, L.; Keitelman, I.A.; Sabbione, F.; Fuentes, F.; Guzman, M.; Galletti, J.G.; Gerber, P.P.; Ostrowski, M.; Geffner, J.R.; Jancic, C.C.; et al. Autophagy Mediates Interleukin-1β Secretion in Human Neutrophils. Front. Immunol. 2018, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Shenderov, K.; Huang, N.-N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of Autophagy by Inflammatory Signals Limits IL-1β Production by Targeting Ubiquitinated Inflammasomes for Destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.R.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between Host Defence, Immune Modulation, and Tissue Injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef]
- Son, S.; Yoon, S.-H.; Chae, B.J.; Hwang, I.; Shim, D.-W.; Choe, Y.H.; Hyun, Y.-M.; Yu, J.-W. Neutrophils Facilitate Prolonged Inflammasome Response in the DAMP-Rich Inflammatory Milieu. Front. Immunol. 2021, 12, 746032. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. TLR Signaling Pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Abdolmaleki, F.; Farahani, N.; Gheibi Hayat, S.M.; Pirro, M.; Bianconi, V.; Barreto, G.E.; Sahebkar, A. The Role of Efferocytosis in Autoimmune Diseases. Front. Immunol. 2018, 9, 1645. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.H.; Genest, J.; Gotto, A.M.; Kastelein, J.J.P.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef]
- Li, D.; Yu, J.; Zeng, R.; Zhao, L.; Wan, Z.; Zeng, Z.; Cao, Y. Neutrophil Count Is Associated With Risks of Cardiovascular Diseases. J. Am. Coll. Cardiol. 2017, 70, 911–912. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.-S.; et al. Hyperglycemia Promotes Myelopoiesis and Impairs the Resolution of Atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as Regulators of Cardiovascular Inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]
- Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int. J. Mol. Sci. 2021, 22, 559. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil Extracellular Trap–Associated Protein Activation of the NLRP3 Inflammasome Is Enhanced in Lupus Macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.S.; Randall, K.L.; Pettitt, J.A.; Ellyard, J.I.; Blumenthal, A.; Enders, A.; Quah, B.J.; Bopp, T.; Parish, C.R.; Brüstle, A. Neutrophil Extracellular Traps and Their Histones Promote Th17 Cell Differentiation Directly via TLR2. Nat. Commun. 2022, 13, 528. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic Stem Cell Release Is Regulated by Circadian Oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef]
- Jiang, K.; Tu, Z.; Chen, K.; Xu, Y.; Chen, F.; Xu, S.; Shi, T.; Qian, J.; Shen, L.; Hwa, J.; et al. Gasdermin D Inhibition Confers Antineutrophil-Mediated Cardioprotection in Acute Myocardial Infarction. J. Clin. Investig. 2022, 132, e151268. [Google Scholar] [CrossRef]
- Gong, Y.; Koh, D.-R. Neutrophils Promote Inflammatory Angiogenesis via Release of Preformed VEGF in an in Vivo Corneal Model. Cell Tissue Res. 2010, 339, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Vafadarnejad, E.; Rizzo, G.; Krampert, L.; Arampatzi, P.; Arias-Loza, A.-P.; Nazzal, Y.; Rizakou, A.; Knochenhauer, T.; Bandi, S.R.; Nugroho, V.A.; et al. Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ. Res. 2020, 127, e232–e249. [Google Scholar] [CrossRef] [PubMed]
- Adrover, J.M.; Del Fresno, C.; Crainiciuc, G.; Cuartero, M.I.; Casanova-Acebes, M.; Weiss, L.A.; Huerga-Encabo, H.; Silvestre-Roig, C.; Rossaint, J.; Cossío, I.; et al. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity 2019, 50, 390–402.e10. [Google Scholar] [CrossRef] [PubMed]
- Aroca-Crevillén, A.; Adrover, J.M.; Hidalgo, A. Circadian Features of Neutrophil Biology. Front. Immunol. 2020, 11, 576. [Google Scholar] [CrossRef]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the Function and Fate of Neutrophils in Sterile Injury and Repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils Orchestrate Post-Myocardial Infarction Healing by Polarizing Macrophages towards a Reparative Phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Conti, P.; Ronconi, G.; Caraffa, A.; Gallenga, C.; Ross, R.; Frydas, I.; Kritas, S. Induction of Pro-Inflammatory Cytokines (IL-1 and IL-6) and Lung Inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-Inflammatory Strategies. J. Biol. Regul. Homeost. Agents 2020, 34, 327–331. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. HLH Across Speciality Collaboration, UK COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet Lond. Engl. 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; de Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes Are Activated in Response to SARS-CoV-2 Infection and Are Associated with COVID-19 Severity in Patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef] [PubMed]
- Rahi, M.S.; Jindal, V.; Reyes, S.-P.; Gunasekaran, K.; Gupta, R.; Jaiyesimi, I. Hematologic Disorders Associated with COVID-19: A Review. Ann. Hematol. 2021, 100, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Zhou, F.; Luo, L.; Xu, M.; Wang, H.; Xia, J.; Gao, Y.; Cai, L.; Wang, Z.; Yin, P.; et al. Haematological Characteristics and Risk Factors in the Classification and Prognosis Evaluation of COVID-19: A Retrospective Cohort Study. Lancet Haematol. 2020, 7, e671–e678. [Google Scholar] [CrossRef] [PubMed]
- Aymonnier, K.; Ng, J.; Fredenburgh, L.E.; Zambrano-Vera, K.; Münzer, P.; Gutch, S.; Fukui, S.; Desjardins, M.; Subramaniam, M.; Baron, R.M.; et al. Inflammasome Activation in Neutrophils of Patients with Severe COVID-19. Blood Adv. 2022, 6, 2001–2013. [Google Scholar] [CrossRef]
- Sánchez-Cerrillo, I.; Landete, P.; Aldave, B.; Sánchez-Alonso, S.; Sánchez-Azofra, A.; Marcos-Jiménez, A.; Ávalos, E.; Alcaraz-Serna, A.; de los Santos, I.; Mateu-Albero, T.; et al. COVID-19 Severity Associates with Pulmonary Redistribution of CD1c+ DCs and Inflammatory Transitional and Nonclassical Monocytes. J. Clin. Investig. 2020, 130, 6290–6300. [Google Scholar] [CrossRef]
- Lakschevitz, F.S.; Hassanpour, S.; Rubin, A.; Fine, N.; Sun, C.; Glogauer, M. Identification of Neutrophil Surface Marker Changes in Health and Inflammation Using High-Throughput Screening Flow Cytometry. Exp. Cell Res. 2016, 342, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, R.; He, G. Hematological Findings in Coronavirus Disease 2019: Indications of Progression of Disease. Ann. Hematol. 2020, 99, 1421–1428. [Google Scholar] [CrossRef]
- Henry, B.M.; de Oliveira, M.H.S.; Benoit, S.; Plebani, M.; Lippi, G. Hematologic, Biochemical and Immune Biomarker Abnormalities Associated with Severe Illness and Mortality in Coronavirus Disease 2019 (COVID-19): A Meta-Analysis. Clin. Chem. Lab. Med. 2020, 58, 1021–1028. [Google Scholar] [CrossRef]
- Junqueira, C.; Crespo, Â.; Ranjbar, S.; Lewandrowski, M.; Ingber, J.; de Lacerda, L.B.; Parry, B.; Ravid, S.; Clark, S.; Ho, F.; et al. SARS-CoV-2 Infects Blood Monocytes to Activate NLRP3 and AIM2 Inflammasomes, Pyroptosis and Cytokine Release. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Beun, R.; Kusadasi, N.; Sikma, M.; Westerink, J.; Huisman, A. Thromboembolic Events and Apparent Heparin Resistance in Patients Infected with SARS-CoV-2. Int. J. Lab. Hematol. 2020, 42 (Suppl. S1), 19–20. [Google Scholar] [CrossRef]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.-D.; Sacco, C.; Bertuzzi, A.; et al. Venous and Arterial Thromboembolic Complications in COVID-19 Patients Admitted to an Academic Hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef]
- Campbell, C.M.; Kahwash, R. Will Complement Inhibition Be the New Target in Treating COVID-19–Related Systemic Thrombosis? Circulation 2020, 141, 1739–1741. [Google Scholar] [CrossRef]
- Tian, S.; Hu, W.; Niu, L.; Liu, H.; Xu, H.; Xiao, S.-Y. Pulmonary Pathology of Early-Phase 2019 Novel Coronavirus (COVID-19) Pneumonia in Two Patients With Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 700–704. [Google Scholar] [CrossRef]
- Laridan, E.; Martinod, K.; De Meyer, S. Neutrophil Extracellular Traps in Arterial and Venous Thrombosis. Semin. Thromb. Hemost. 2019, 45, 86–93. [Google Scholar] [CrossRef]
- Münzer, P.; Negro, R.; Fukui, S.; di Meglio, L.; Aymonnier, K.; Chu, L.; Cherpokova, D.; Gutch, S.; Sorvillo, N.; Shi, L.; et al. NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions. Front. Immunol. 2021, 12, 683803. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.Y.; Al-Omran, M.; Stewart, D.J.; et al. Angiotensin Converting Enzyme-2 Confers Endothelial Protection and Attenuates Atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1377–H1384. [Google Scholar] [CrossRef] [PubMed]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA Traps Promote Thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, P.; Wei, Y.; Yue, H.; Wang, Y.; Hu, M.; Zhang, S.; Cao, T.; Yang, C.; Li, M.; et al. Histopathologic Changes and SARS-CoV-2 Immunostaining in the Lung of a Patient With COVID-19. Ann. Intern. Med. 2020, 172, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 Forms an IL-1beta-Processing Inflammasome with Increased Activity in Muckle-Wells Autoinflammatory Disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Fenini, G.; Contassot, E.; French, L.E. Potential of IL-1, IL-18 and Inflammasome Inhibition for the Treatment of Inflammatory Skin Diseases. Front. Pharmacol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a New Gene Encoding a Putative Pyrin-like Protein Causes Familial Cold Autoinflammatory Syndrome and Muckle-Wells Syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Neven, B.; Callebaut, I.; Prieur, A.-M.; Feldmann, J.; Bodemer, C.; Lepore, L.; Derfalvi, B.; Benjaponpitak, S.; Vesely, R.; Sauvain, M.J.; et al. Molecular Basis of the Spectral Expression of CIAS1 Mutations Associated with Phagocytic Cell-Mediated Autoinflammatory Disorders CINCA/NOMID, MWS, and FCU. Blood 2004, 103, 2809–2815. [Google Scholar] [CrossRef] [PubMed]
- Brydges, S.D.; Mueller, J.L.; McGeough, M.D.; Pena, C.A.; Misaghi, A.; Gandhi, C.; Putnam, C.D.; Boyle, D.L.; Firestein, G.S.; Horner, A.A.; et al. Inflammasome-Mediated Disease Animal Models Reveal Roles for Innate but Not Adaptive Immunity. Immunity 2009, 30, 875–887. [Google Scholar] [CrossRef]
- Kool, M.; Pétrilli, V.; De Smedt, T.; Rolaz, A.; Hammad, H.; van Nimwegen, M.; Bergen, I.M.; Castillo, R.; Lambrecht, B.N.; Tschopp, J. Cutting Edge: Alum Adjuvant Stimulates Inflammatory Dendritic Cells through Activation of the NALP3 Inflammasome. J. Immunol. Baltim. Md 1950 2008, 181, 3755–3759. [Google Scholar] [CrossRef]
- Camilli, G.; Bohm, M.; Piffer, A.C.; Lavenir, R.; Williams, D.L.; Neven, B.; Grateau, G.; Georgin-Lavialle, S.; Quintin, J. β-Glucan-Induced Reprogramming of Human Macrophages Inhibits NLRP3 Inflammasome Activation in Cryopyrinopathies. J. Clin. Investig. 2020, 130, 4561–4573. [Google Scholar] [CrossRef]
- Ley, K.; Hoffman, H.M.; Kubes, P.; Cassatella, M.A.; Zychlinsky, A.; Hedrick, C.C.; Catz, S.D. Neutrophils: New Insights and Open Questions. Sci. Immunol. 2018, 3, eaat4579. [Google Scholar] [CrossRef]
- Hoffman, H.M.; Wanderer, A.A.; Broide, D.H. Familial Cold Autoinflammatory Syndrome: Phenotype and Genotype of an Autosomal Dominant Periodic Fever. J. Allergy Clin. Immunol. 2001, 108, 615–620. [Google Scholar] [CrossRef]
- Stackowicz, J.; Gaudenzio, N.; Serhan, N.; Conde, E.; Godon, O.; Marichal, T.; Starkl, P.; Balbino, B.; Roers, A.; Bruhns, P.; et al. Neutrophil-Specific Gain-of-Function Mutations in Nlrp3 Promote Development of Cryopyrin-Associated Periodic Syndrome. J. Exp. Med. 2021, 218, e20201466. [Google Scholar] [CrossRef]
- Németh, T.; Sperandio, M.; Mócsai, A. Neutrophils as Emerging Therapeutic Targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef]
- O’Byrne, P.M.; Metev, H.; Puu, M.; Richter, K.; Keen, C.; Uddin, M.; Larsson, B.; Cullberg, M.; Nair, P. Efficacy and Safety of a CXCR2 Antagonist, AZD5069, in Patients with Uncontrolled Persistent Asthma: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Respir. Med. 2016, 4, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Challagundla, N.; Saha, B.; Agrawal-Rajput, R. Insights into Inflammasome Regulation: Cellular, Molecular, and Pathogenic Control of Inflammasome Activation. Immunol. Res. 2022, 70, 578–606. [Google Scholar] [CrossRef]
- Dalbeth, N.; Gosling, A.L.; Gaffo, A.; Abhishek, A. Gout. Lancet Lond. Engl. 2021, 397, 1843–1855. [Google Scholar] [CrossRef]
- Delaleu, J.; Lepelletier, C.; Calugareanu, A.; De Masson, A.; Charvet, E.; Petit, A.; Giurgea, I.; Amselem, S.; Karabina, S.; Jachiet, M.; et al. Neutrophilic Dermatoses. Rev. Med. Interne 2022, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwaniuk, A.; Jablonska, E. Neutrophils in Health and Disease: From Receptor Sensing to Inflammasome Activation. Int. J. Mol. Sci. 2023, 24, 6340. https://doi.org/10.3390/ijms24076340
Iwaniuk A, Jablonska E. Neutrophils in Health and Disease: From Receptor Sensing to Inflammasome Activation. International Journal of Molecular Sciences. 2023; 24(7):6340. https://doi.org/10.3390/ijms24076340
Chicago/Turabian StyleIwaniuk, Agnieszka, and Ewa Jablonska. 2023. "Neutrophils in Health and Disease: From Receptor Sensing to Inflammasome Activation" International Journal of Molecular Sciences 24, no. 7: 6340. https://doi.org/10.3390/ijms24076340
APA StyleIwaniuk, A., & Jablonska, E. (2023). Neutrophils in Health and Disease: From Receptor Sensing to Inflammasome Activation. International Journal of Molecular Sciences, 24(7), 6340. https://doi.org/10.3390/ijms24076340