
Engineering a HER2-CAR-NK Cell Secreting Soluble Programmed Cell Death Protein with Superior Antitumor Efficacy
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
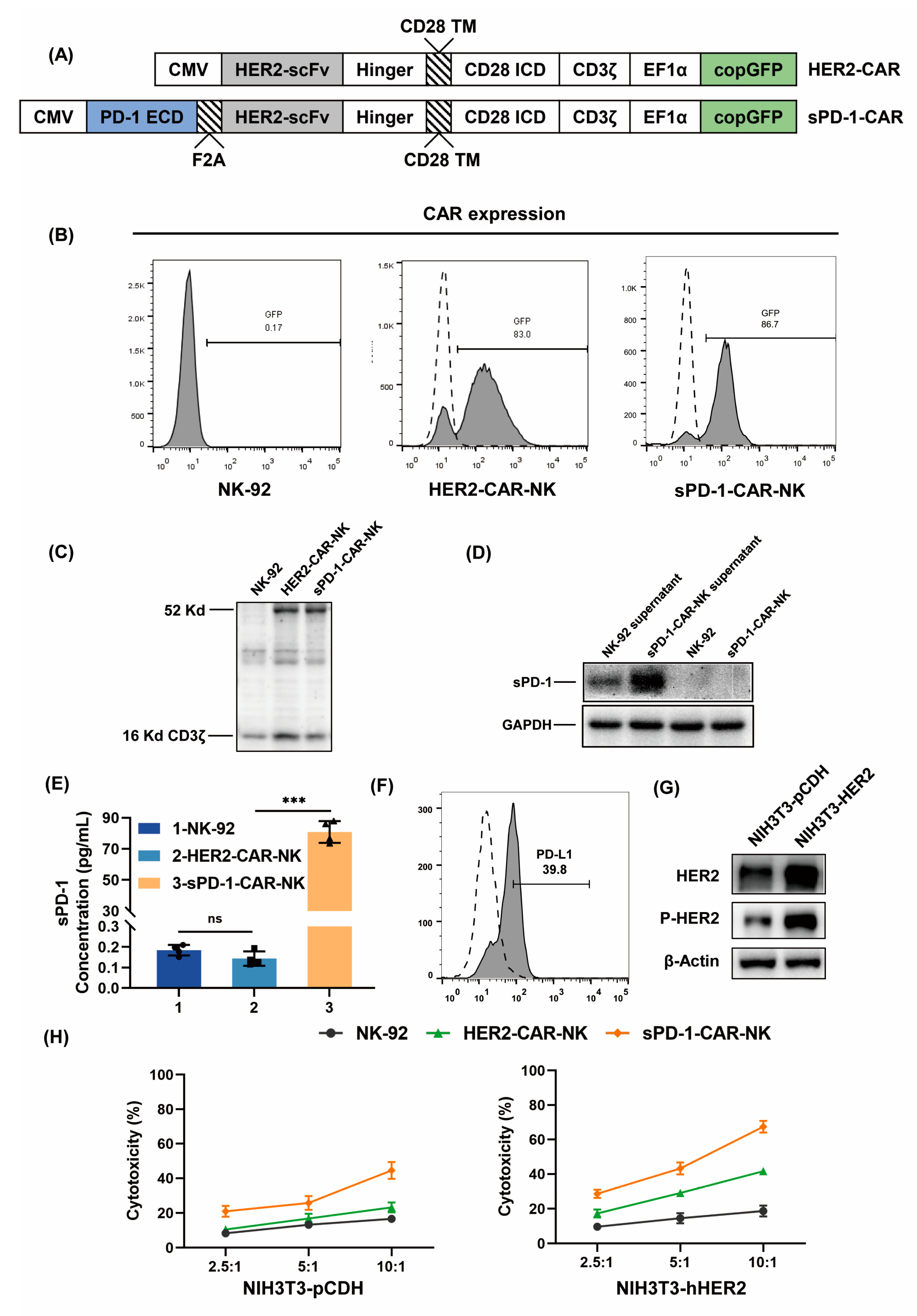
2.1. Generation of HER2-CAR-NK Cells and sPD-1-CAR-NK Cells
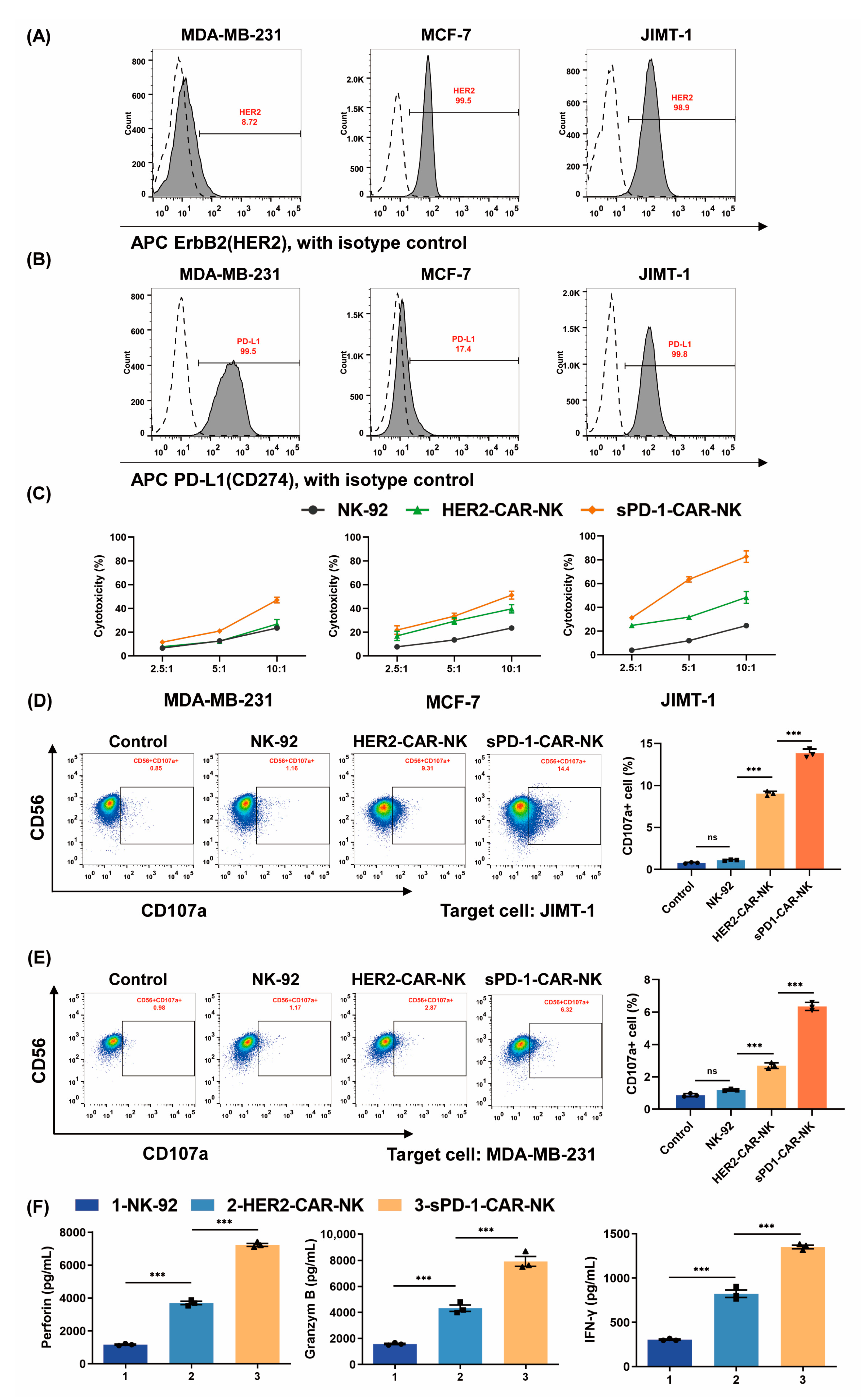
2.2. sPD-1-CAR-NK Cells Enhance Cytotoxicity toward HER2- and PD-L1-Positive Breast Cancer Cells
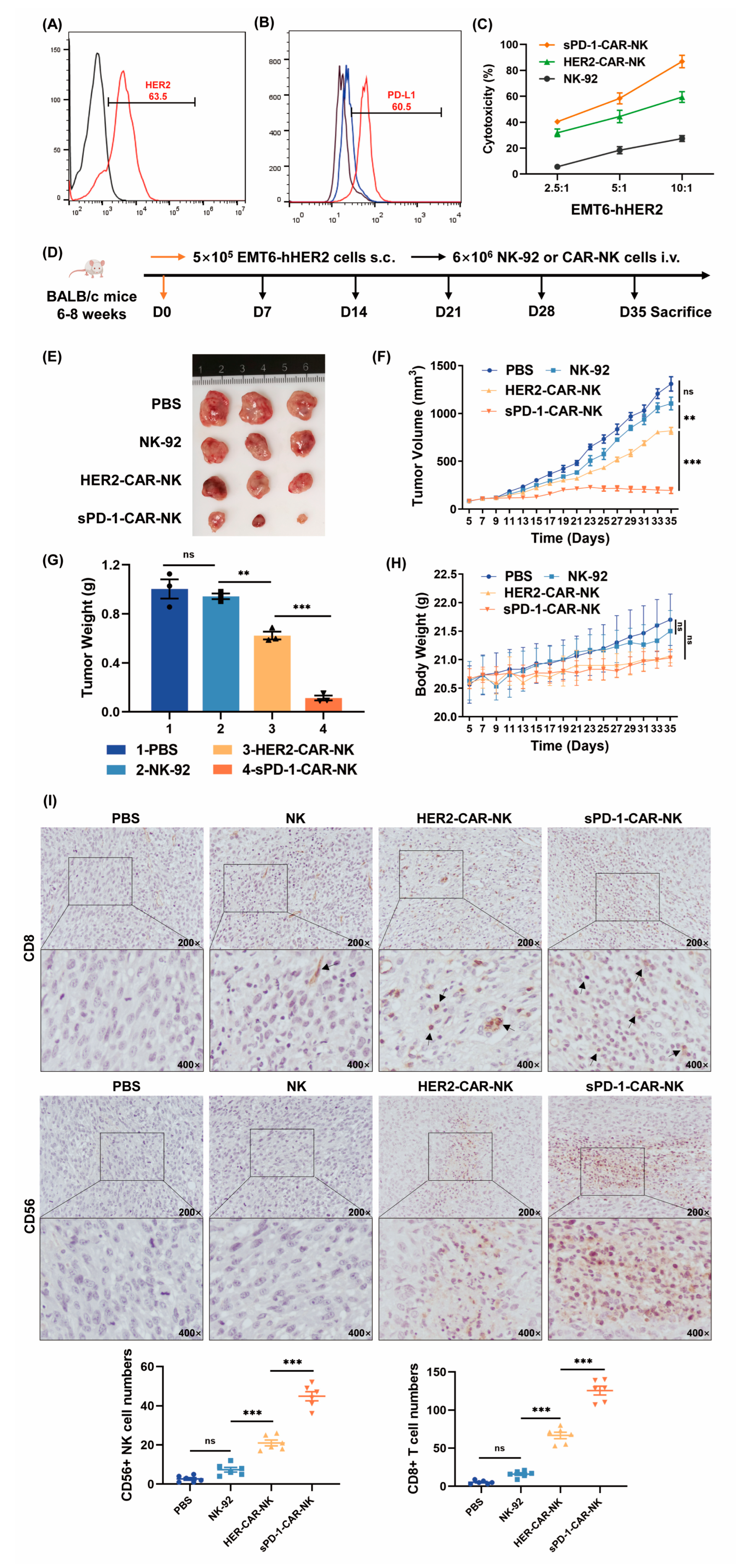
2.3. sPD-1-CAR-NK Cells More Effectively Inhibit Growth of Target Humanized hHER2-EMT6 Xenografts
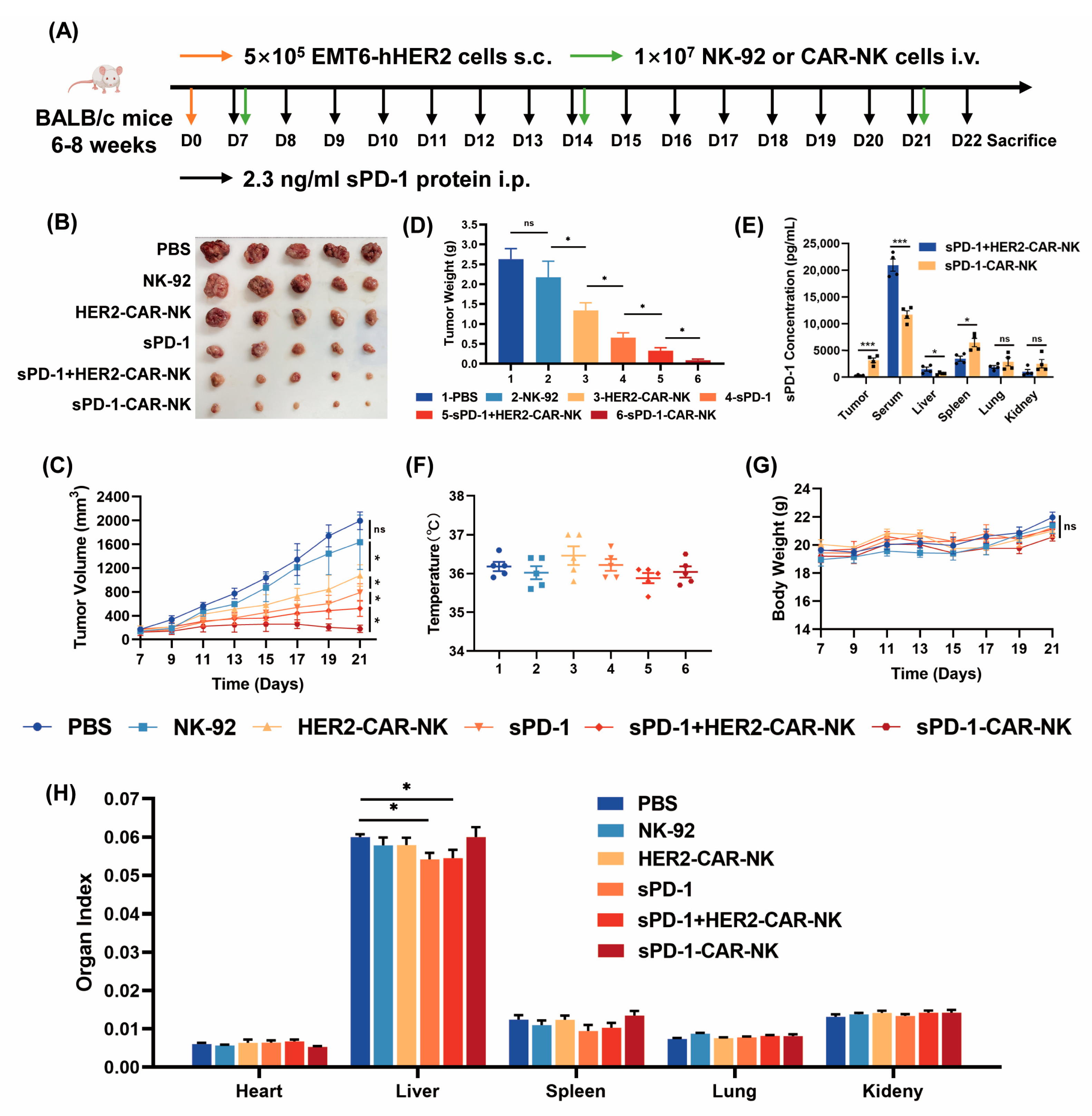
2.4. Co-Expression of sPD-1 with HER2-CAR in NK-92 Cells Has Antitumor Efficacy Superior to HER2-CAR Plus sPD-1
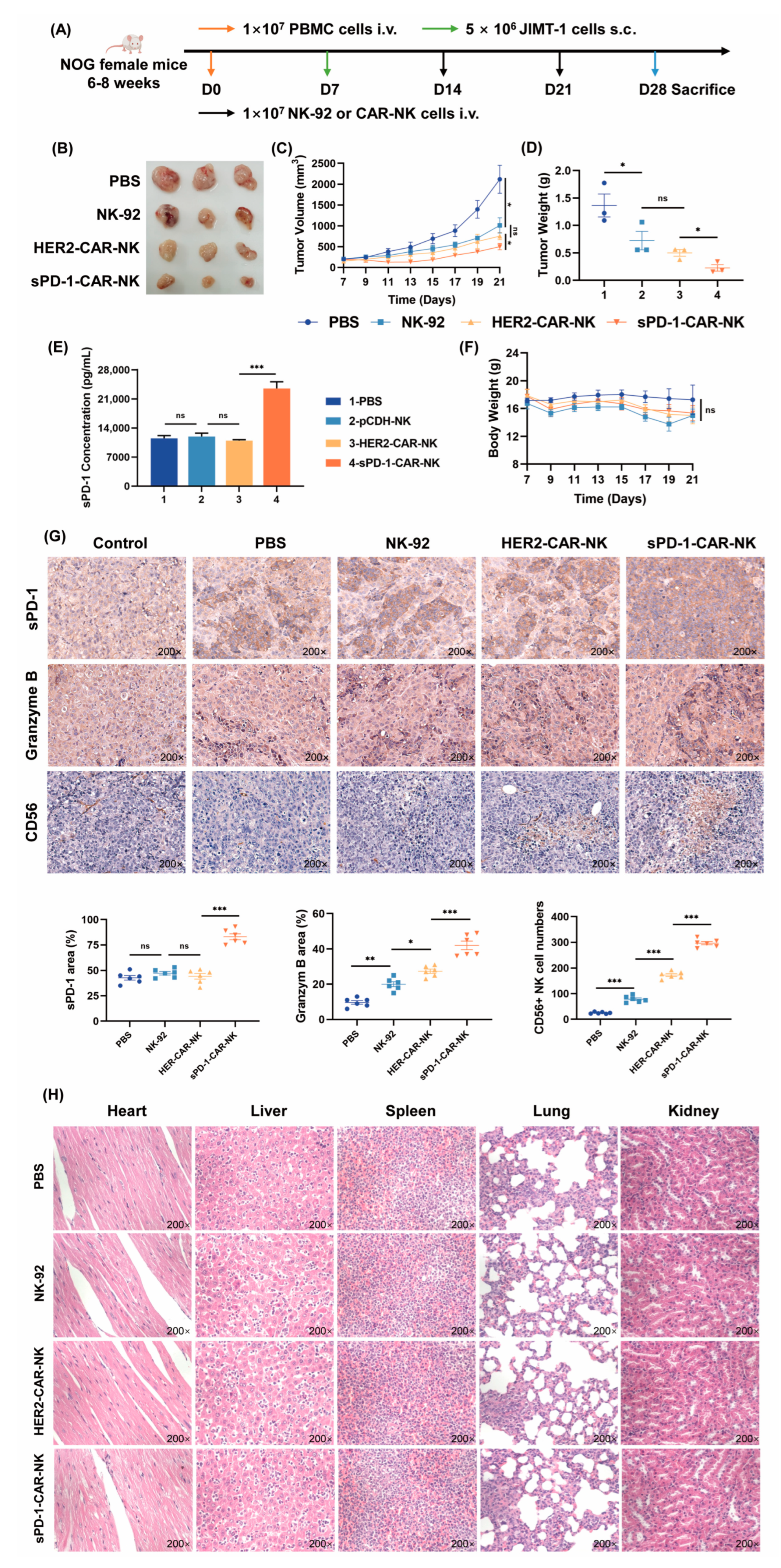
2.5. sPD-1-CAR-NK Cells More Effectively Block the Growth of JIMT-1 Cell Xenografts in Immune Humanized NOG Mice
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture Conditions
4.2. Isolation and Purification of PBMC
4.3. Construction of Lentiviral CAR-Expression Vector
4.4. Lentivirus Production
4.5. Generation of HER2-CAR-NK Cells and sPD-1-CAR-NK Cells
4.6. Flow Cytometry Analysis
4.7. Analysis of Cytokine and sPD-1 Protein Levels by ELISA
4.8. Western Blot Analysis
4.9. Cytotoxicity Assay
4.10. Mouse Xenograft Studies
4.11. Immunohistochemistry
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gingras, I.; Gebhart, G.; de Azambuja, E.; Piccart-Gebhart, M. HER2-Positive Breast Cancer Is Lost in Translation: Time for Patient-Centered Research. Nat. Rev. Clin. Oncol. 2017, 14, 669–681. [Google Scholar] [CrossRef]
- Goutsouliak, K.; Veeraraghavan, J.; Sethunath, V.; De Angelis, C.; Osborne, C.K.; Rimawi, M.F.; Schiff, R. Towards Personalized Treatment for Early Stage HER2-Positive Breast Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 233–250. [Google Scholar] [CrossRef]
- Wang, J.; Xu, B. Targeted Therapeutic Options and Future Perspectives for HER2-Positive Breast Cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Schlam, I.; Tarantino, P.; Tolaney, S.M. Overcoming Resistance to HER2-Directed Therapies in Breast Cancer. Cancers 2022, 14, 3996. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the Tumor Microenvironment in PD-L1/PD-1-Mediated Tumor Immune Escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The Diverse Functions of the PD1 Inhibitory Pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 Expression in the Tumor Microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, M.; Liu, Y.; Yi, M.; Jiao, D.; Wu, K. Biological Characteristics and Clinical Significance of Soluble PD-1/PD-L1 and Exosomal PD-L1 in Cancer. Front. Immunol. 2022, 13, 827921. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-Y.; Park, S.-H.; Nam, H.J.; Choi, D.-H.; Sung, Y.-C. Enhancement of Vaccine-Induced Primary and Memory CD8(+) T-Cell Responses by Soluble PD-1. J. Immunother. Hagerstown 2011, 34, 297–306. [Google Scholar] [CrossRef]
- Wei, J.; Luo, C.; Wang, Y.; Guo, Y.; Dai, H.; Tong, C.; Ti, D.; Wu, Z.; Han, W. PD-1 Silencing Impairs the Anti-Tumor Function of Chimeric Antigen Receptor Modified T Cells by Inhibiting Proliferation Activity. J. Immunother. Cancer 2019, 7, 209. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted Delivery of a PD-1-Blocking ScFv by CAR-T Cells Enhances Anti-Tumor Efficacy in Vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Nakajima, M.; Sakoda, Y.; Adachi, K.; Nagano, H.; Tamada, K. Improved Survival of Chimeric Antigen Receptor-Engineered T (CAR-T) and Tumor-Specific T Cells Caused by Anti-Programmed Cell Death Protein 1 Single-Chain Variable Fragment-Producing CAR-T Cells. Cancer Sci. 2019, 110, 3079–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, D.; Ao, X.; Yang, Y.; Chen, Z.; Xu, X. Soluble Immune Checkpoints in Cancer: Production, Function and Biological Significance. J. Immunother. Cancer 2018, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.-F.; Hsu, P.-N. Cancer Immunotherapy by Targeting Immune Checkpoints: Mechanism of T Cell Dysfunction in Cancer Immunity and New Therapeutic Targets. J. Biomed. Sci. 2017, 24, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, N.; Porter, D. Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol. Blood Marrow Transplant. 2019, 25, e123–e127. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Klein Wolterink, R.G.J.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric Antigen Receptor Natural Killer (CAR-NK) Cell Design and Engineering for Cancer Therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef]
- Siegler, E.L.; Zhu, Y.; Wang, P.; Yang, L. Off-the-Shelf CAR-NK Cells for Cancer Immunotherapy. Cell Stem Cell 2018, 23, 160–161. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective Inhibition of Tumor Growth by Clonal NK Cells Expressing an ErbB2/HER2-Specific Chimeric Antigen Receptor. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budi, H.S.; Ahmad, F.N.; Achmad, H.; Ansari, M.J.; Mikhailova, M.V.; Suksatan, W.; Chupradit, S.; Shomali, N.; Marofi, F. Human Epidermal Growth Factor Receptor 2 (HER2)-Specific Chimeric Antigen Receptor (CAR) for Tumor Immunotherapy; Recent Progress. Stem Cell Res. Ther. 2022, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Quatrini, L.; Mariotti, F.R.; Munari, E.; Tumino, N.; Vacca, P.; Moretta, L. The Immune Checkpoint PD-1 in Natural Killer Cells: Expression, Function and Targeting in Tumour Immunotherapy. Cancers 2020, 12, 3285. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L.; Yu, G.; Phillips, J.H. Co-Association of CD3 Zeta with a Receptor (CD16) for IgG Fc on Human Natural Killer Cells. Nature 1989, 342, 803–805. [Google Scholar] [CrossRef]
- Deswal, S.; Schamel, W.W.A. CD3ζ. In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer: New York, NY, USA, 2012; pp. 306–313. ISBN 978-1-4419-0461-4. [Google Scholar]
- Wang, X.-H.; Guo, W.; Qiu, W.; Ao, L.-Q.; Yao, M.-W.; Xing, W.; Yu, Y.; Chen, Q.; Wu, X.-F.; Li, Z.; et al. Fibroblast-like Cells Promote Wound Healing via PD-L1-Mediated Inflammation Resolution. Int. J. Biol. Sci. 2022, 18, 4388–4399. [Google Scholar] [CrossRef] [PubMed]
- Amaral, I.; Silva, C.; Correia-Branco, A.; Martel, F. Effect of Metformin on Estrogen and Progesterone Receptor-Positive (MCF-7) and Triple-Negative (MDA-MB-231) Breast Cancer Cells. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 102, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Tanner, M.; Kapanen, A.I.; Junttila, T.; Raheem, O.; Grenman, S.; Elo, J.; Elenius, K.; Isola, J. Characterization of a Novel Cell Line Established from a Patient with Herceptin-Resistant Breast Cancer. Mol. Cancer Ther. 2004, 3, 1585–1592. [Google Scholar] [CrossRef]
- Blatteis, C.M.; Sehic, E. Cytokines and Fever. Ann. N. Y. Acad. Sci. 1998, 840, 608–618. [Google Scholar] [CrossRef]
- Daher, M.; Rezvani, K. Outlook for New CAR-Based Therapies with a Focus on CAR NK Cells: What Lies beyond CAR-Engineered T Cells in the Race against Cancer. Cancer Discov. 2021, 11, 45–58. [Google Scholar] [CrossRef]
- Liu, H.; Yang, B.; Sun, T.; Lin, L.; Hu, Y.; Deng, M.; Yang, J.; Liu, T.; Li, J.; Sun, S.; et al. Specific Growth Inhibition of ErbB2-expressing Human Breast Cancer Cells by Genetically Modified NK-92 Cells. Oncol. Rep. 2015, 33, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Poznanski, S.M.; Ritchie, T.M.; Fan, I.Y.; El-Sayes, A.; Portillo, A.L.; Ben-Avi, R.; Rojas, E.A.; Chew, M.V.; Shargall, Y.; Ashkar, A.A. Expanded Human NK Cells from Lung Cancer Patients Sensitize Patients’ PDL1-Negative Tumors to PD1-Blockade Therapy. J. Immunother. Cancer 2021, 9, e001933. [Google Scholar] [CrossRef] [PubMed]
- Hasim, M.S.; Marotel, M.; Hodgins, J.J.; Vulpis, E.; Makinson, O.J.; Asif, S.; Shih, H.-Y.; Scheer, A.K.; MacMillan, O.; Alonso, F.G.; et al. When Killers Become Thieves: Trogocytosed PD-1 Inhibits NK Cells in Cancer. Sci. Adv. 2022, 8, eabj3286. [Google Scholar] [CrossRef]
- Zhu, X.; Lang, J. Soluble PD-1 and PD-L1: Predictive and Prognostic Significance in Cancer. Oncotarget 2017, 8, 97671–97682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, C.; Ohm-Laursen, L.; Barington, T.; Husby, S.; Lillevang, S.T. Alternative Splice Variants of the Human PD-1 Gene. Cell. Immunol. 2005, 235, 109–116. [Google Scholar] [CrossRef]
- Ruan, Y.; Hu, W.; Li, W.; Lu, H.; Gu, H.; Zhang, Y.; Zhu, C.; Chen, Q. Analysis of Plasma EBV-DNA and Soluble Checkpoint Proteins in Nasopharyngeal Carcinoma Patients after Definitive Intensity-Modulated Radiotherapy. BioMed Res. Int. 2019, 2019, 3939720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasikumar, P.; Shrimali, R.; Adurthi, S.; Ramachandra, R.; Satyam, L.; Dhudashiya, A.; Samiulla, D.; Sunilkumar, K.B.; Ramachandra, M. A Novel Peptide Therapeutic Targeting PD1 Immune Checkpoint with Equipotent Antagonism of Both Ligands and a Potential for Better Management of Immune-Related Adverse Events. J. Immunother. Cancer 2013, 1, O24. [Google Scholar] [CrossRef] [Green Version]
- Maki, G.; Klingemann, H.G.; Martinson, J.A.; Tam, Y.K. Factors Regulating the Cytotoxic Activity of the Human Natural Killer Cell Line, NK-92. J. Hematother. Stem Cell Res. 2001, 10, 369–383. [Google Scholar] [CrossRef]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and Safety of Trastuzumab as a Single Agent in First-Line Treatment of HER2-Overexpressing Metastatic Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef]
- Kenanova, V.; Olafsen, T.; Crow, D.M.; Sundaresan, G.; Subbarayan, M.; Carter, N.H.; Ikle, D.N.; Yazaki, P.J.; Chatziioannou, A.F.; Gambhir, S.S.; et al. Tailoring the Pharmacokinetics and Positron Emission Tomography Imaging Properties of Anti-Carcinoembryonic Antigen Single-Chain Fv-Fc Antibody Fragments. Cancer Res. 2005, 65, 622–631. [Google Scholar] [CrossRef]
- Seyedmirzaei, H.; Keshavarz-Fathi, M.; Razi, S.; Gity, M.; Rezaei, N. Recent Progress in Immunotherapy of Breast Cancer Targeting the Human Epidermal Growth Factor Receptor 2 (HER2). J. Oncol. Pharm. Pract. 2021, 27, 1235–1244. [Google Scholar] [CrossRef]
- Wartewig, T.; Kurgyis, Z.; Keppler, S.; Pechloff, K.; Hameister, E.; Öllinger, R.; Maresch, R.; Buch, T.; Steiger, K.; Winter, C.; et al. PD-1 Is a Haploinsufficient Suppressor of T Cell Lymphomagenesis. Nature 2017, 552, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Wang, X.-J.; Chen, D.-X.; Liu, X.-N.; Wang, X.-J. Humanized Mouse Model: A Review on Preclinical Applications for Cancer Immunotherapy. Am. J. Cancer Res. 2020, 10, 4568–4584. [Google Scholar]
- Crouse, J.; Xu, H.C.; Lang, P.A.; Oxenius, A. NK Cells Regulating T Cell Responses: Mechanisms and Outcome. Trends Immunol. 2015, 36, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Zen, Y.; Yeh, M.M. Hepatotoxicity of Immune Checkpoint Inhibitors: A Histology Study of Seven Cases in Comparison with Autoimmune Hepatitis and Idiosyncratic Drug-Induced Liver Injury. Mod. Pathol. 2018, 31, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, W.; Chen, J.; Hou, W.; Chen, J.; Xiong, Y.; Li, H.; Qi, X.; Xu, H.; Xie, Z.; Li, M.; et al. Engineering a HER2-CAR-NK Cell Secreting Soluble Programmed Cell Death Protein with Superior Antitumor Efficacy. Int. J. Mol. Sci. 2023, 24, 6843. https://doi.org/10.3390/ijms24076843
Xia W, Chen J, Hou W, Chen J, Xiong Y, Li H, Qi X, Xu H, Xie Z, Li M, et al. Engineering a HER2-CAR-NK Cell Secreting Soluble Programmed Cell Death Protein with Superior Antitumor Efficacy. International Journal of Molecular Sciences. 2023; 24(7):6843. https://doi.org/10.3390/ijms24076843
Chicago/Turabian StyleXia, Wenjiao, Jiaxin Chen, Wenqing Hou, Junsheng Chen, Ying Xiong, Hongyan Li, Xin Qi, Hui Xu, Zuoquan Xie, Mingfeng Li, and et al. 2023. "Engineering a HER2-CAR-NK Cell Secreting Soluble Programmed Cell Death Protein with Superior Antitumor Efficacy" International Journal of Molecular Sciences 24, no. 7: 6843. https://doi.org/10.3390/ijms24076843
APA StyleXia, W., Chen, J., Hou, W., Chen, J., Xiong, Y., Li, H., Qi, X., Xu, H., Xie, Z., Li, M., Zhang, X., & Li, J. (2023). Engineering a HER2-CAR-NK Cell Secreting Soluble Programmed Cell Death Protein with Superior Antitumor Efficacy. International Journal of Molecular Sciences, 24(7), 6843. https://doi.org/10.3390/ijms24076843