
Oxime-Linked Peptide–Daunomycin Conjugates as Good Tools for Selection of Suitable Homing Devices in Targeted Tumor Therapy: An Overview

 , , , , ,
, , , , ,  ,
,
Abstract
:1. Introduction
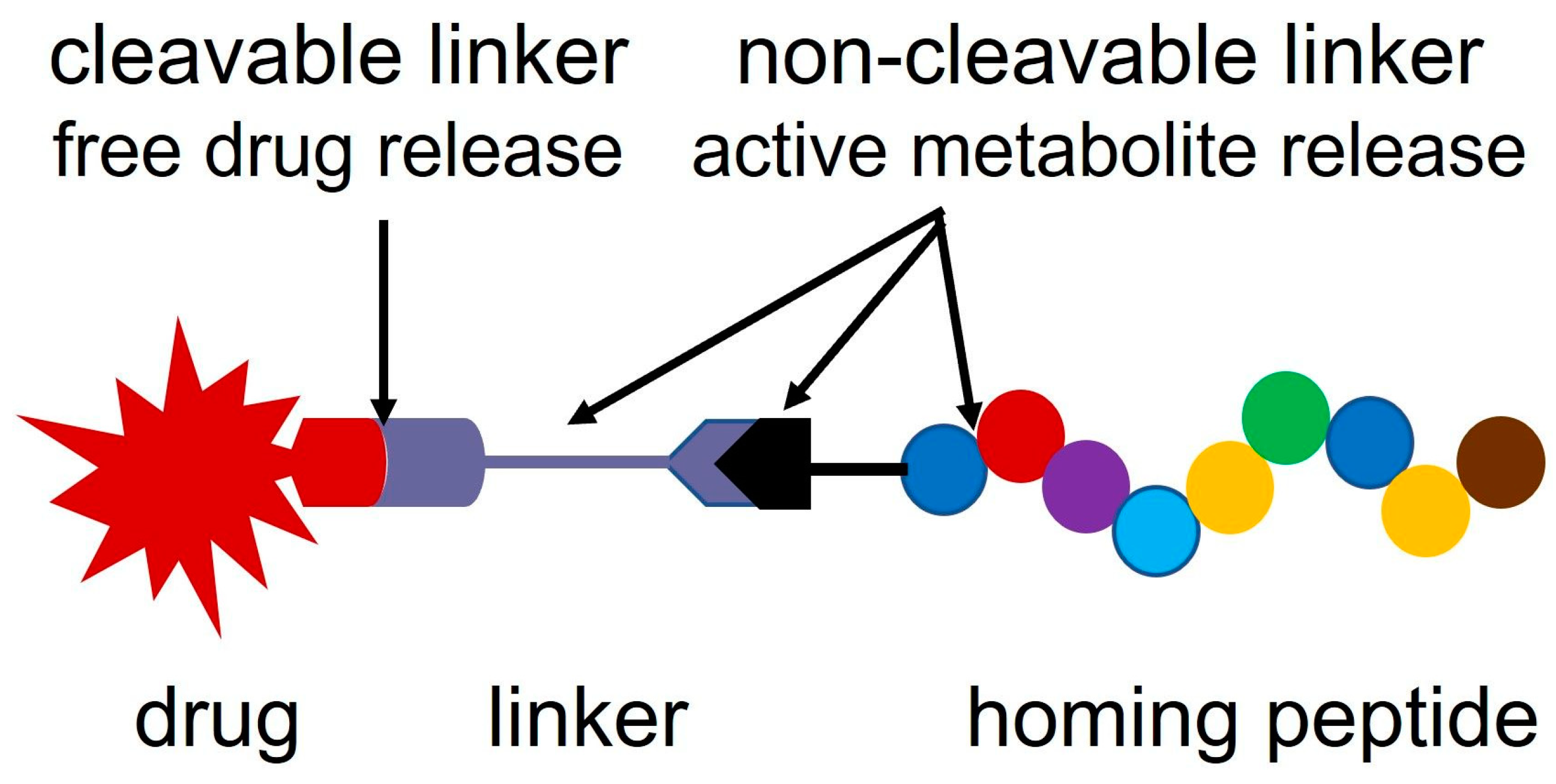
2. Linker Systems
2.1. Ester Linkage
2.2. Hydrazone Linkage
2.3. Self-Immolative Linkers
2.4. Disulfide Bond
2.5. Amide Bond
2.6. Thioether Linkage
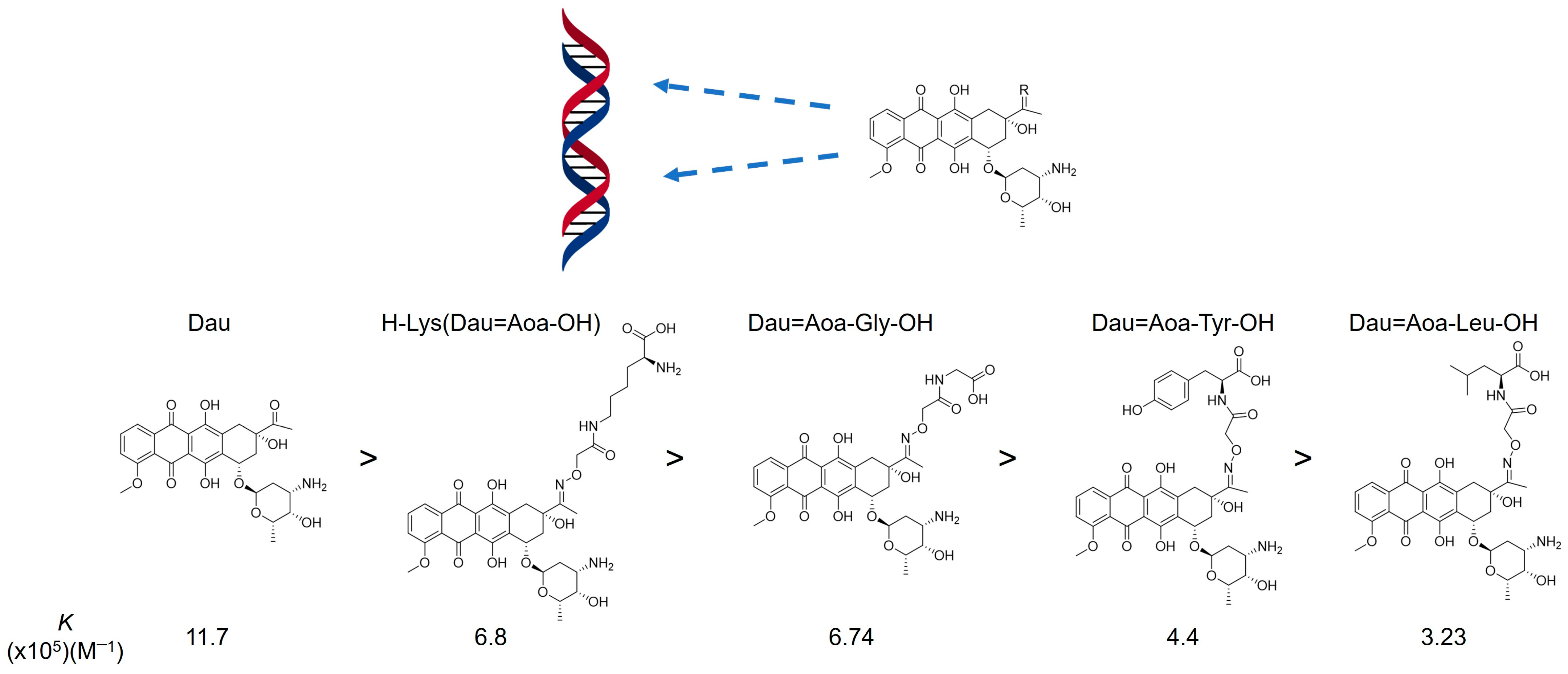
2.7. Oxime Linkage
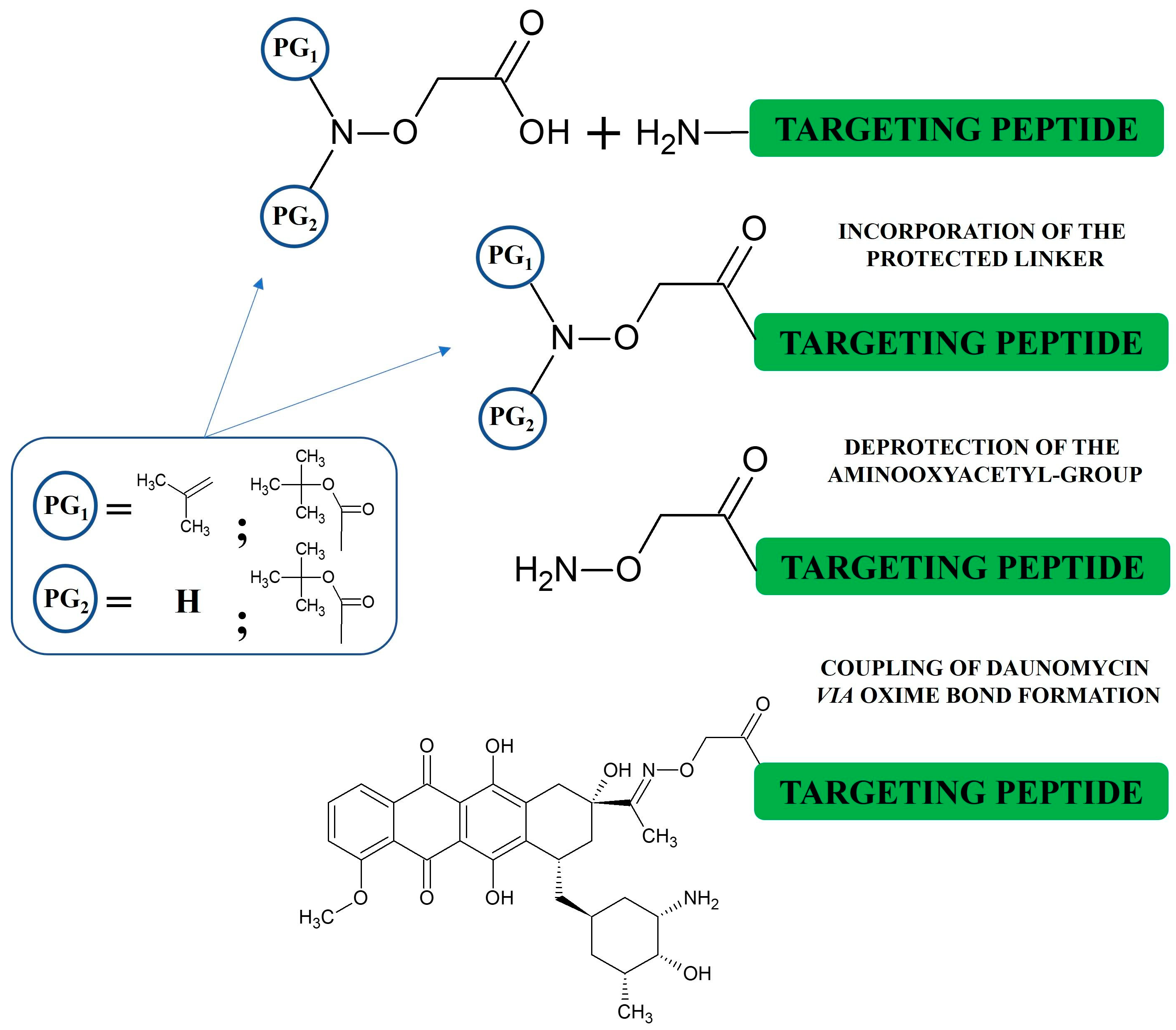
3. Synthesis of Oxime-Linked Peptide–Drug Conjugates
4. Development of Oxime-Linked Peptide–Daunomycin Conjugate
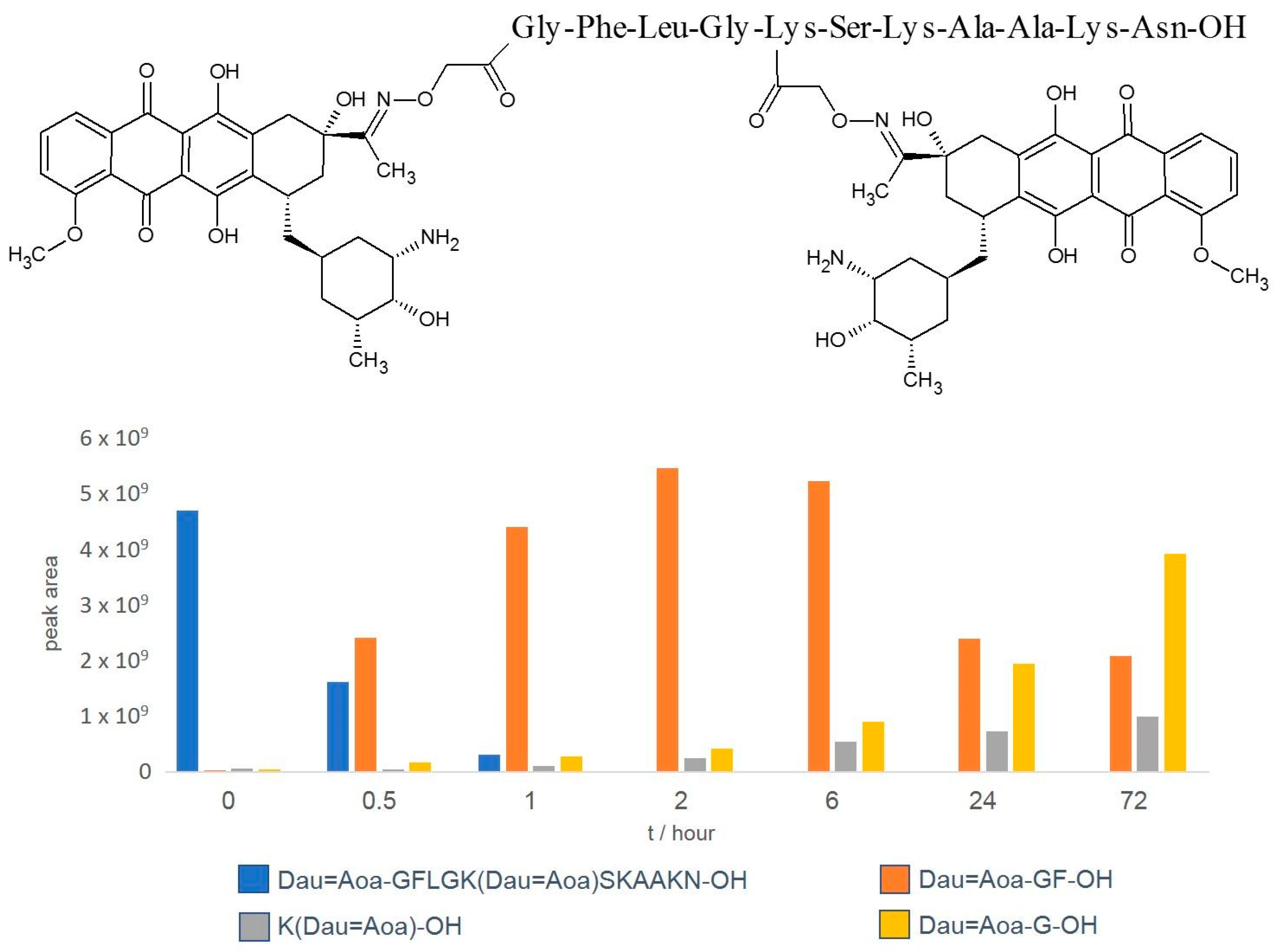
4.1. Degradation of Oxime-Linked Peptide–Drug Conjugates in the Presence of Cathepsin B or Lysosomal Homogenate
4.2. Cardiotoxicity of Oxime-Linked Anthracycline–Peptide Conjugates
4.3. Application of Oxime-Linked Peptide–Daunomycin Conjugates for the Development of Appropriate Drug Delivery Systems
4.4. In Vivo Acute and Chronic Toxicity of Oxime-Linked Daunomycin–Peptide Conjugates
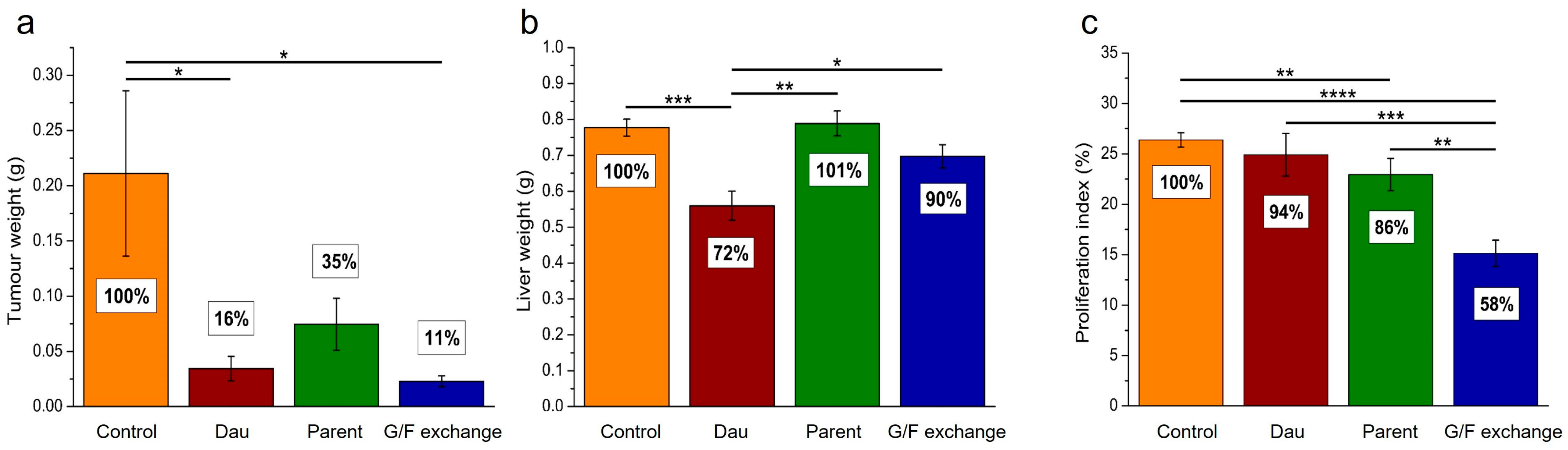
4.5. In Vivo Tumor Growth Inhibition with Oxime-Linked Daunomycin–Peptide Conjugates
4.6. In Vivo Antitumor Effect of Oxime-Linked GnRH-III Derivative–Dau Conjugates
5. Development of Bombesin-Based Peptide–Drug Conjugates
6. Improved Antitumor Activity of Selected Homing Peptide from Phage Display Library by Sequence Optimization
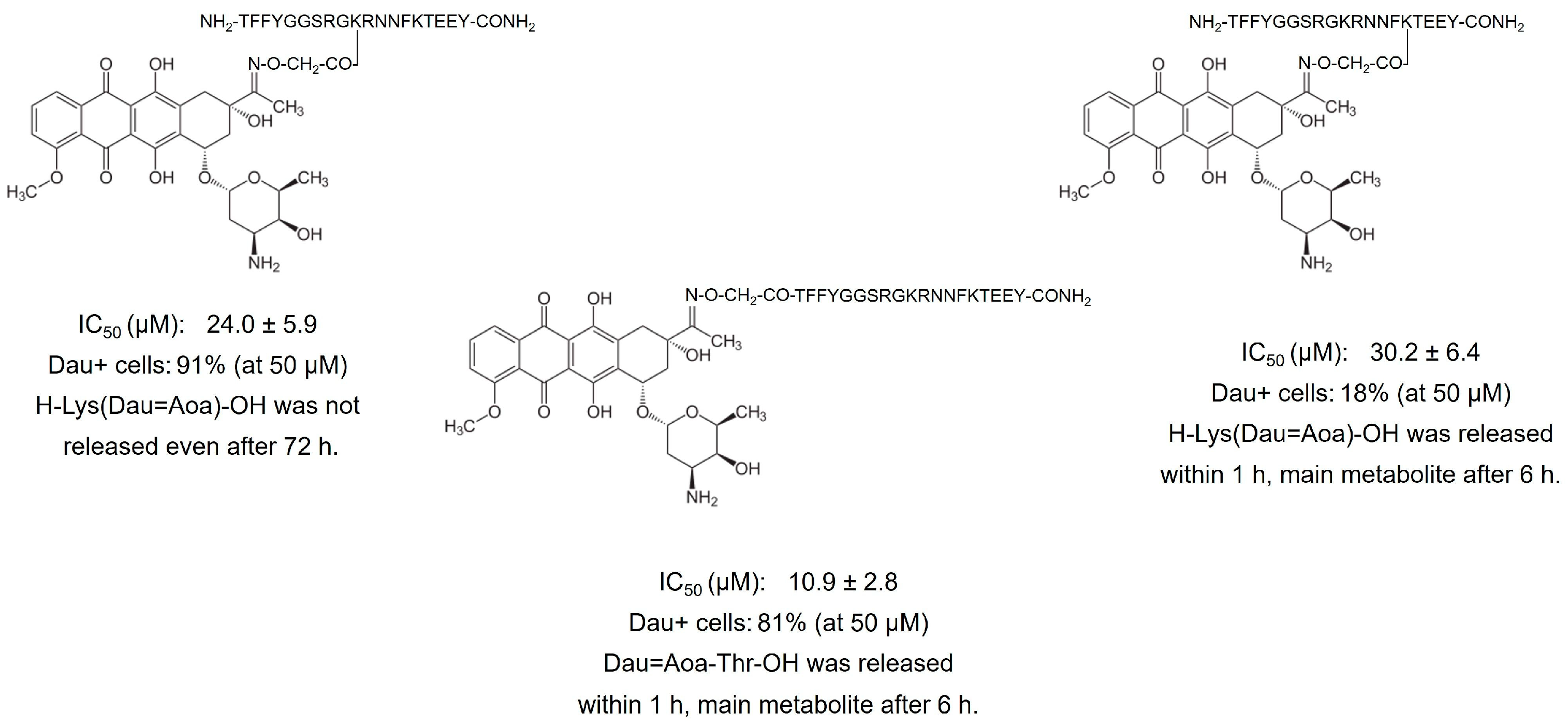
6.1. Phage Display-Based Homing Peptide Linked to Daunomycin via Oxime Bond for Selective Drug Targeting of HT-29 Colon Cancer
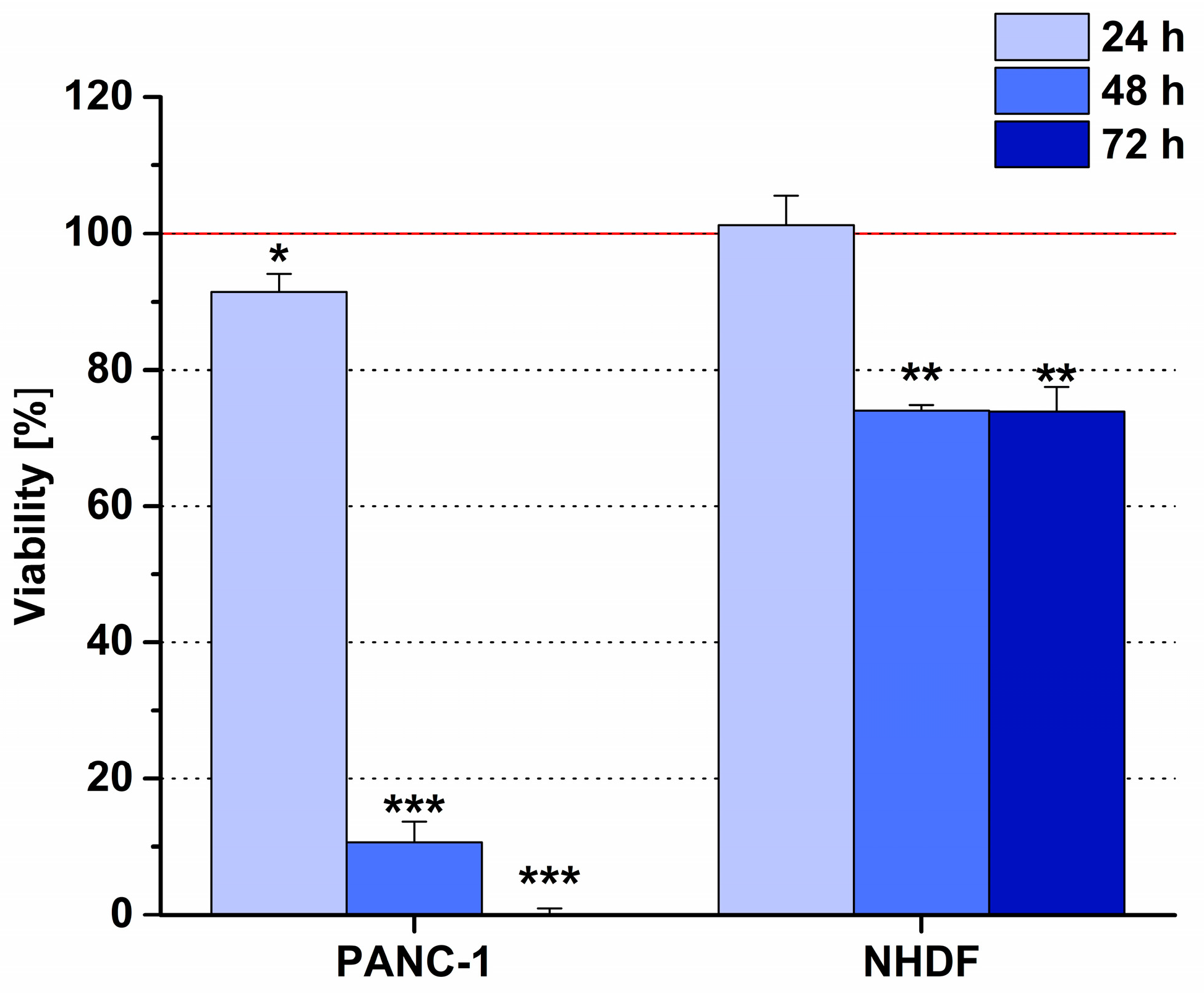
6.2. Phage Display-Based Homing Peptide Linked to Daunomycin via Oxime Bond for Selective Drug Targeting of PANC-1 Pancreatic Cancer
7. Other Examples of Oxime-Based Conjugates for Targeted Tumor Therapy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Correction Statement
References
- World Health Organization. GLOBOCAN 2020. Available online: https://Gco.Iarc.Fr/Today/Home (accessed on 3 February 2022).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Zhu, Y.-S.; Tang, K.; Lv, J. Peptide–Drug Conjugate-Based Novel Molecular Drug Delivery System in Cancer. Trends Pharmacol. Sci. 2021, 42, 857–869. [Google Scholar] [CrossRef]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide–Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef]
- Wadhawan, A.; Chatterjee, M.; Singh, G. Present Scenario of Bioconjugates in Cancer Therapy: A Review. Int. J. Mol. Sci. 2019, 20, 5243. [Google Scholar] [CrossRef]
- Cilurzo, F.; Cristiano, M.C.; Da Pian, M.; Cianflone, E.; Quintieri, L.; Paolino, D.; Pasut, G. Overcoming Cancer Cell Drug Resistance by a Folic Acid Targeted Polymeric Conjugate of Buthionine Sulfoximine. Anticancer Agents Med. Chem. 2019, 19, 1513–1522. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Vrettos, E.I.; Mező, G.; Tzakos, A.G. On the Design Principles of Peptide–Drug Conjugates for Targeted Drug Delivery to the Malignant Tumor Site. Beilstein J. Org. Chem. 2018, 14, 930–954. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Guan, X.; Ma, H.; Cong, H.; Zhang, W.; Miao, Z. Small Molecule-Drug Conjugates: A Novel Strategy for Cancer-Targeted Treatment. Eur. J. Med. Chem. 2019, 163, 883–895. [Google Scholar] [CrossRef]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, Y.; Li, W.; Jeanty, C.; Xiang, G.; Dong, Y. Recent Advances of Antibody Drug Conjugates for Clinical Applications. Acta Pharm. Sin. B 2020, 10, 1589–1600. [Google Scholar] [CrossRef]
- Heh, E.; Allen, J.; Ramirez, F.; Lovasz, D.; Fernandez, L.; Hogg, T.; Riva, H.; Holland, N.; Chacon, J. Peptide Drug Conjugates and Their Role in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 829. [Google Scholar] [CrossRef]
- Chavda, V.P.; Solanki, H.K.; Davidson, M.; Apostolopoulos, V.; Bojarska, J. Peptide-Drug Conjugates: A New Hope for Cancer Management. Molecules 2022, 27, 7232. [Google Scholar] [CrossRef]
- Ghaly, H.S.A.; Varamini, P. New Drug Delivery Strategies Targeting the GnRH Receptor in Breast and Other Cancers. Endocr. Relat. Cancer 2021, 28, R251–R269. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.; Li, M.; Wu, X.; Setrerrahmane, S.; Xu, H. Integrins as Attractive Targets for Cancer Therapeutics. Acta Pharm. Sin. B 2021, 11, 2726–2737. [Google Scholar] [CrossRef] [PubMed]
- Le Joncour, V.; Laakkonen, P. Seek & Destroy, Use of Targeting Peptides for Cancer Detection and Drug Delivery. Bioorganic Med. Chem. 2018, 26, 2797–2806. [Google Scholar] [CrossRef]
- Lingasamy, P.; Teesalu, T. Homing Peptides for Cancer Therapy. In Bio-Nanomedicine for Cancer Therapy; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2021; Volume 1295, pp. 29–48. [Google Scholar] [CrossRef]
- Kiss, K.; Biri-Kovács, B.; Szabó, R.; Ranđelović, I.; Enyedi, K.N.; Schlosser, G.; Orosz, Á.; Kapuvári, B.; Tóvári, J.; Mező, G. Sequence Modification of Heptapeptide Selected by Phage Display as Homing Device for HT-29 Colon Cancer Cells to Improve the Anti-Tumour Activity of Drug Delivery Systems. Eur. J. Med. Chem. 2019, 176, 105–116. [Google Scholar] [CrossRef]
- Schuster, S.; Juhász, É.; Halmos, G.; Neundorf, I.; Gennari, C.; Mező, G. Development and Biochemical Characterization of Self-Immolative Linker Containing GnRH-III-Drug Conjugates. Int. J. Mol. Sci. 2022, 23, 5071. [Google Scholar] [CrossRef]
- Szabó, I.; Manea, M.; Orbán, E.; Csámpai, A.; Bősze, S.; Szabó, R.; Tejeda, M.; Gaál, D.; Kapuvári, B.; Przybylski, M.; et al. Development of an Oxime Bond Containing Daunorubicin-Gonadotropin-Releasing Hormone-III Conjugate as a Potential Anticancer Drug. Bioconjug. Chem. 2009, 20, 656–665. [Google Scholar] [CrossRef]
- Chatzisideri, T.; Leonidis, G.; Sarli, V. Cancer-Targeted Delivery Systems Based on Peptides. Future Med. Chem. 2018, 10, 2201–2226. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.F.A.; Abbas, N.; Zhang, H.; Fang, Q. Identification of a PH-Responsive Peptide–Paclitaxel Conjugate as a Novel Drug with Improved Therapeutic Potential. J. Med. Chem. 2023, 66, 8324–8337. [Google Scholar] [CrossRef]
- Sayyad, N.; Vrettos, E.I.; Karampelas, T.; Chatzigiannis, C.M.; Spyridaki, K.; Liapakis, G.; Tamvakopoulos, C.; Tzakos, A.G. Development of Bioactive Gemcitabine-D-Lys6-GnRH Prodrugs with Linker-Controllable Drug Release Rate and Enhanced Biopharmaceutical Profile. Eur. J. Med. Chem. 2019, 166, 256–266. [Google Scholar] [CrossRef]
- Wang, L.; Chen, H.; Wang, F.; Zhang, X. The Development of Peptide-Drug Conjugates (PDCs) Strategies for Paclitaxel. Expert Opin. Drug Deliv. 2022, 19, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tian, Y.; Jiang, S.; Wang, Z. A Novel Homodimer Peptide–Drug Conjugate Improves the Efficacy of HER2-Positive Breast Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 4590. [Google Scholar] [CrossRef] [PubMed]
- Ziaei, E.; Saghaeidehkordi, A.; Dill, C.; Maslennikov, I.; Chen, S.; Kaur, K. Targeting Triple Negative Breast Cancer Cells with Novel Cytotoxic Peptide–Doxorubicin Conjugates. Bioconjugate Chem. 2019, 30, 3098–3106. [Google Scholar] [CrossRef] [PubMed]
- Saghaeidehkordi, A.; Chen, S.; Yang, S.; Kaur, K. Evaluation of a Keratin 1 Targeting Peptide-Doxorubicin Conjugate in a Mouse Model of Triple-Negative Breast Cancer. Pharmaceutics 2021, 13, 661. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Guo, R.; Xuan, M.; Sun, Q.; Wu, W. An Acid-Sensitive Nanofiber Conjugate Based on a Short Aromatic Peptide for Targeted Delivery of Doxorubicin in Liver Cancer. Int. J. Nanomed. 2022, 17, 2961–2973. [Google Scholar] [CrossRef]
- Gonzaga, R.V.; do Nascimento, L.A.; Santos, S.S.; Machado Sanches, B.A.; Giarolla, J.; Ferreira, E.I. Perspectives About Self-Immolative Drug Delivery Systems. J. Pharm. Sci. 2020, 109, 3262–3281. [Google Scholar] [CrossRef]
- Pryyma, A.; Matinkhoo, K.; Bu, Y.J.; Merkens, H.; Zhang, Z.; Bénard, F.; Perrin, D.M. Synthesis and Preliminary Evaluation of Octreotate Conjugates of Bioactive Synthetic Amatoxins for Targeting Somatostatin Receptor (Sstr2) Expressing Cells. RSC Chem. Biol. 2022, 3, 69–78. [Google Scholar] [CrossRef]
- Borbély, A.; Figueras, E.; Martins, A.; Esposito, S.; Auciello, G.; Monteagudo, E.; Di Marco, A.; Summa, V.; Cordella, P.; Perego, R.; et al. Synthesis and Biological Evaluation of RGD–Cryptophycin Conjugates for Targeted Drug Delivery. Pharmaceutics 2019, 11, 151. [Google Scholar] [CrossRef]
- Böhme, D.; Beck-Sickinger, A.G. Controlling Toxicity of Peptide–Drug Conjugates by Different Chemical Linker Structures. ChemMedChem 2015, 10, 804–814. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Cao, J.; Meng, Q.; Li, X.; Zhang, Y.; Lam, K.S.; Hong, A.; Liu, R.; Chen, X. Development and Characterization of a Novel Peptide—Drug Conjugate with DM1 for Treatment of FGFR2-Positive Tumors. Biomedicines 2021, 9, 849. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Hu, J.; Liu, S. Disulfide-Based Self-Immolative Linkers and Functional Bioconjugates for Biological Applications. Macromol. Rapid Commun. 2020, 41, 1900531. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Li, S.; Wang, X.; Zhang, Y.; Sun, Y.; Wang, Y.; Wang, X.; He, B.; Dai, W.; Zhang, H.; et al. A Comparative Study of the Antitumor Efficacy of Peptide-Doxorubicin Conjugates with Different Linkers. J. Control. Release 2018, 275, 129–141. [Google Scholar] [CrossRef]
- Dirisala, A.; Uchida, S.; Tockary, T.A.; Yoshinaga, N.; Li, J.; Osawa, S.; Gorantla, L.; Fukushima, S.; Osada, K.; Kataoka, K. Precise Tuning of Disulphide Crosslinking in MRNA Polyplex Micelles for Optimising Extracellular and Intracellular Nuclease Tolerability. J. Drug Target. 2019, 27, 670–680. [Google Scholar] [CrossRef]
- Xie, J.; Gonzalez-Carter, D.; Tockary, T.A.; Nakamura, N.; Xue, Y.; Nakakido, M.; Akiba, H.; Dirisala, A.; Liu, X.; Toh, K.; et al. Dual-Sensitive Nanomicelles Enhancing Systemic Delivery of Therapeutically Active Antibodies Specifically into the Brain. ACS Nano 2020, 14, 6729–6742. [Google Scholar] [CrossRef]
- Krishnan, M.A.; Sengupta, S.; Chelvam, V. Preparation of Ligand-Targeted Drug Conjugates for Cancer Therapy and Their Evaluation In Vitro. Curr. Protoc. Chem. Biol. 2018, 10, e50. [Google Scholar] [CrossRef] [PubMed]
- Spears, R.J.; McMahon, C.; Chudasama, V. Cysteine Protecting Groups: Applications in Peptide and Protein Science. Chem. Soc. Rev. 2021, 50, 11098–11155. [Google Scholar] [CrossRef]
- Almuqbil, R.M.; Heyder, R.S.; Bielski, E.R.; Durymanov, M.; Reineke, J.J.; da Rocha, S.R.P. Dendrimer Conjugation Enhances Tumor Penetration and Efficacy of Doxorubicin in Extracellular Matrix-Expressing 3D Lung Cancer Models. Mol. Pharm. 2020, 17, 1648–1662. [Google Scholar] [CrossRef]
- Böhme, D.; Krieghoff, J.; Beck-Sickinger, A.G. Double Methotrexate-Modified Neuropeptide Y Analogues Express Increased Toxicity and Overcome Drug Resistance in Breast Cancer Cells. J. Med. Chem. 2016, 59, 3409–3417. [Google Scholar] [CrossRef]
- Gilad, Y.; Noy, E.; Senderowitz, H.; Albeck, A.; Firer, M.A.; Gellerman, G. Dual-drug RGD Conjugates Provide Enhanced Cytotoxicity to Melanoma and Non-small Lung Cancer Cells. Pept. Sci. 2016, 106, 160–171. [Google Scholar] [CrossRef]
- Klimpel, A.; Neundorf, I. Bifunctional Peptide Hybrids Targeting the Matrix of Mitochondria. J. Control. Release 2018, 291, 147–156. [Google Scholar] [CrossRef]
- Kalia, J.; Raines, R.T. Hydrolytic Stability of Hydrazones and Oximes. Angew. Chem. Int. Ed. 2008, 47, 7523–7526. [Google Scholar] [CrossRef] [PubMed]
- Sestak, J.; Mullins, M.; Northrup, L.; Thati, S.; Laird Forrest, M.; Siahaan, T.J.; Berkland, C. Single-Step Grafting of Aminooxy-Peptides to Hyaluronan: A Simple Approach to Multifunctional Therapeutics for Experimental Autoimmune Encephalomyelitis. J. Control. Release 2013, 168, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Edupuganti, O.P.; Renaudet, O.; Defrancq, E.; Dumy, P. The Oxime Bond Formation as an Efficient Chemical Tool for the Preparation of 3′,5′-Bifunctionalised Oligodeoxyribonucleotides. Bioorg. Med. Chem. Lett. 2004, 14, 2839–2842. [Google Scholar] [CrossRef]
- Grigalevicius, S.; Chierici, S.; Renaudet, O.; Lo-Man, R.; Dériaud, E.; Leclerc, C.; Dumy, P. Chemoselective Assembly and Immunological Evaluation of Multiepitopic Glycoconjugates Bearing Clustered Tn Antigen as Synthetic Anticancer Vaccines. Bioconjug. Chem. 2005, 16, 1149–1159. [Google Scholar] [CrossRef]
- Nagy, A.; Schally, A.V.; Armatis, P.; Szepeshazi, K.; Halmos, G.; Kovacs, M.; Zarandi, M.; Groot, K.; Miyazaki, M.; Jungwirth, A.; et al. Cytotoxic Analogs of Luteinizing Hormone-Releasing Hormone Containing Doxorubicin or 2-Pyrrolinodoxorubicin, a Derivative 500–1000 Times More Potent. Proc. Natl. Acad. Sci. USA 1996, 93, 7269–7273. [Google Scholar] [CrossRef]
- Hou, L.; Hou, Y.; Liang, Y.; Chen, B.; Zhang, X.; Wang, Y.; Zhou, K.; Zhong, T.; Long, B.; Pang, W.; et al. Anti-Tumor Effects of P-LPK-CPT, a Peptide-Camptothecin Conjugate, in Colorectal Cancer. Commun. Biol. 2022, 5, 1248. [Google Scholar] [CrossRef]
- Ché, C.; Yang, G.; Thiot, C.; Lacoste, M.-C.; Currie, J.-C.; Demeule, M.; Régina, A.; Béliveau, R.; Castaigne, J.-P. New Angiopep-Modified Doxorubicin (ANG1007) and Etoposide (ANG1009) Chemotherapeutics with Increased Brain Penetration. J. Med. Chem. 2010, 53, 2814–2824. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Gründker, C. The Role of Gonadotropin-Releasing Hormone (GnRH) in Endometrial Cancer. Cells 2021, 10, 292. [Google Scholar] [CrossRef]
- Nagy, A.; Plonowski, A.; Schally, A.V. Stability of Cytotoxic Luteinizing Hormone-Releasing Hormone Conjugate (AN-152) Containing Doxorubicin 14-O-Hemiglutarate in Mouse and Human Serum in Vitro: Implications for the Design of Preclinical Studies. Proc. Natl. Acad. Sci. USA 2000, 97, 829–834. [Google Scholar] [CrossRef]
- Schlage, P.; Mező, G.; Orbán, E.; Bősze, S.; Manea, M. Anthracycline-GnRH Derivative Bioconjugates with Different Linkages: Synthesis, in Vitro Drug Release and Cytostatic Effect. J. Control. Release 2011, 156, 170–178. [Google Scholar] [CrossRef]
- Sleno, L.; Campagna-Slater, V.; Volmer, D.A. Dissociation Reactions of Protonated Anthracycline Antibiotics Following Electrospray Ionization-Tandem Mass Spectrometry. Int. J. Mass. Spectrom. 2006, 255–256, 130–138. [Google Scholar] [CrossRef]
- Pethő, L.; Mező, G.; Schlosser, G. Overcharging Effect in Electrospray Ionization Mass Spectra of Daunomycin-Tuftsin Bioconjugates. Molecules 2019, 24, 2981. [Google Scholar] [CrossRef]
- Peter, S.; Alven, S.; Maseko, R.B.; Aderibigbe, B.A. Doxorubicin-Based Hybrid Compounds as Potential Anticancer Agents: A Review. Molecules 2022, 27, 4478. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable Linkers in Antibody–Drug Conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A.; Frigoli, M.; Prevosti, M.; Mason, M.; Bucci, R.; Belvisi, L.; Pignataro, L.; Gennari, C. Advanced Pyrrolidine-Carbamate Self-Immolative Spacer with Tertiary Amine Handle Induces Superfast Cyclative Drug Release. ChemMedChem 2022, 17, e202200279. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Gund, M.G.; Desai, D.C.; Borhade, N.; Senthilkumar, S.P.; Dhiman, M.; Mangu, N.K.; Mali, S.V.; Dubash, N.P.; Halder, S.; et al. Mutual Prodrugs Containing Bio-Cleavable and Drug Releasable Disulfide Linkers. Bioorganic Chem. 2013, 49, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guan, J.; Wan, J.; Li, Z. Disulfide Based Prodrugs for Cancer Therapy. RSC Adv. 2020, 10, 24397–24409. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhao, H.; Huang, G.; Liu, J.; He, W.; Huang, Q. ES-MION-Based Dual-Modality PET/MRI Probes for Acidic Tumor Microenvironment Imaging. ACS Omega 2022, 7, 3442–3451. [Google Scholar] [CrossRef]
- Pershina, A.G.; Demin, A.M.; Perekucha, N.A.; Brikunova, O.Y.; Efimova, L.V.; Nevskaya, K.V.; Vakhrushev, A.V.; Zgoda, V.G.; Uimin, M.A.; Minin, A.S.; et al. Peptide Ligands on the PEGylated Nanoparticle Surface and Human Serum Composition Are Key Factors for the Interaction between Immune Cells and Nanoparticles. Colloids Surf. B Biointerfaces 2023, 221, 112981. [Google Scholar] [CrossRef]
- Rose, K. Facile Synthesis of Homogeneous Artificial Proteins. J. Am. Chem. Soc. 1994, 116, 30–33. [Google Scholar] [CrossRef]
- Decostaire, I.P.; Lelièvre, D.; Zhang, H.; Delmas, A.F. Controlling the Outcome of Overacylation of N-Protected Aminooxyacetic Acid during the Synthesis of an Aminooxy-Peptide for Chemical Ligation. Tetrahedron Lett. 2006, 47, 7057–7060. [Google Scholar] [CrossRef]
- Jiménez-Castells, C.; de la Torre, B.G.; Gutiérrez Gallego, R.; Andreu, D. Optimized Synthesis of Aminooxy-Peptides as Glycoprobe Precursors for Surface-Based Sugar–Protein Interaction Studies. Bioorg. Med. Chem. Lett. 2007, 17, 5155–5158. [Google Scholar] [CrossRef]
- Foillard, S.; Rasmussen, M.O.; Razkin, J.; Boturyn, D.; Dumy, P. 1-Ethoxyethylidene, a New Group for the Stepwise SPPS of Aminooxyacetic Acid Containing Peptides. J. Org. Chem. 2008, 73, 983–991. [Google Scholar] [CrossRef]
- Cai, R.Z.; Szoke, B.; Lu, R.; Fu, D.; Redding, T.W.; Schally, A. V Synthesis and Biological Activity of Highly Potent Octapeptide Analogs of Somatostatin. Proc. Natl. Acad. Sci. USA 1986, 83, 1896–1900. [Google Scholar] [CrossRef] [PubMed]
- Mezö, G.; Szabó, I.; Kertész, I.; Hegedüs, R.; Orbán, E.; Leurs, U.; Bösze, S.; Halmos, G.; Manea, M. Efficient Synthesis of an (Aminooxy) Acetylated-somatostatin Derivative Using (Aminooxy)Acetic Acid as a ‘Carbonyl Capture’ Reagent. J. Pept. Sci. 2011, 17, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Enyedi, K.N.; Tóth, S.; Szakács, G.; Mező, G. NGR-Peptide−drug Conjugates with Dual Targeting Properties. PLoS ONE 2017, 12, e0178632. [Google Scholar] [CrossRef] [PubMed]
- Ingallinella, P.; Di Marco, A.; Taliani, M.; Fattori, D.; Pessi, A. A New Method for Chemoselective Conjugation of Unprotected Peptides to Dauno- and Doxorubicin. Bioorg. Med. Chem. Lett. 2001, 11, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.; Sloane, B.F. Cathepsin B: Multiple Roles in Cancer. Proteom. Clin. Appl. 2014, 8, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Orbán, E.; Mező, G.; Schlage, P.; Csík, G.; Kulić, Ž.; Ansorge, P.; Fellinger, E.; Möller, H.M.; Manea, M. In Vitro Degradation and Antitumor Activity of Oxime Bond-Linked Daunorubicin–GnRH-III Bioconjugates and DNA-Binding Properties of Daunorubicin–Amino Acid Metabolites. Amino Acids 2011, 41, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Hegedüs, R.; Pauschert, A.; Orbán, E.; Szabó, I.; Andreu, D.; Marquardt, A.; Mező, G.; Manea, M. Modification of Daunorubicin-GnRH-III Bioconjugates with Oligoethylene Glycol Derivatives to Improve Solubility and Bioavailability for Targeted Cancer Chemotherapy. Pept. Sci. 2015, 104, 167–177. [Google Scholar] [CrossRef]
- Zhang, J.; Cui, X.; Yan, Y.; Li, M.; Yang, Y.; Wang, J.; Zhang, J. Research Progress of Cardioprotective Agents for Prevention of Anthracycline Cardiotoxicity. Am. J. Transl. Res. 2016, 8, 2862–2875. [Google Scholar]
- Polgár, L.; Lajkó, E.; Soós, P.; Láng, O.; Manea, M.; Merkely, B.; Mező, G.; Kőhidai, L. Drug Targeting to Decrease Cardiotoxicity—Determination of the Cytotoxic Effect of GnRH-Based Conjugates Containing Doxorubicin, Daunorubicin and Methotrexate on Human Cardiomyocytes and Endothelial Cells. Beilstein J. Org. Chem. 2018, 14, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Régina, A.; Ché, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.-P.; Béliveau, R. Identification and Design of Peptides as a New Drug Delivery System for the Brain. J. Pharmacol. Exp. Ther. 2008, 324, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Régina, A.; Demeule, M.; Ché, C.; Lavallée, I.; Poirier, J.; Gabathuler, R.; Béliveau, R.; Castaigne, J. Antitumour Activity of ANG1005, a Conjugate between Paclitaxel and the New Brain Delivery Vector Angiopep-2. Br. J. Pharmacol. 2008, 155, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Pethő, L.; Oláh-Szabó, R.; Mező, G. Influence of the Drug Position on Bioactivity in Angiopep-2—Daunomycin Conjugates. Int. J. Mol. Sci. 2023, 24, 3106. [Google Scholar] [CrossRef] [PubMed]
- Sower, S.A.; Chiang, Y.C.; Lovas, S.; Conlon, J.M. Primary Structure and Biological Activity of a Third Gonadotropin-Releasing Hormone from Lamprey Brain. Endocrinology 1993, 132, 1125–1131. [Google Scholar] [CrossRef]
- Lovas, S.; Páyi, I.; Vincze, B.; Horvath, J.; Kovács, M.; Mezö, I.; Tóth, G.; Tepán, I.; Murphy, R.F. Direct Anticancer Activity of Gonadotropin-releasing Hormone-III. J. Pept. Res. 1998, 52, 384–389. [Google Scholar] [CrossRef]
- Manea, M.; Tóvári, J.; Tejeda, M.; Schulcz, Á.; Kapuvári, B.; Vincze, B.; Mező, G. In-Vivo Antitumour Effect of Daunorubicin–GnRH-III Derivative Conjugates on Colon Carcinoma-Bearing Mice. Anticancer Drugs 2012, 23, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Kapuvári, B.; Hegedüs, R.; Schulcz, Á.; Manea, M.; Tóvári, J.; Gacs, A.; Vincze, B.; Mező, G. Improved in Vivo Antitumor Effect of a Daunorubicin—GnRH-III Bioconjugate Modified by Apoptosis Inducing Agent Butyric Acid on Colorectal Carcinoma Bearing Mice. Investig. New Drugs 2016, 34, 416–423. [Google Scholar] [CrossRef]
- Ranđelović, I.; Schuster, S.; Kapuvári, B.; Fossati, G.; Steinkühler, C.; Mező, G.; Tóvári, J. Improved In Vivo Anti-Tumor and Anti-Metastatic Effect of GnRH-III-Daunorubicin Analogs on Colorectal and Breast Carcinoma Bearing Mice. Int. J. Mol. Sci. 2019, 20, 4763. [Google Scholar] [CrossRef] [PubMed]
- Gomena, J.; Vári, B.; Oláh-Szabó, R.; Biri-Kovács, B.; Bősze, S.; Borbély, A.; Soós, Á.; Ranđelović, I.; Tóvári, J.; Mező, G. Targeting the Gastrin-Releasing Peptide Receptor (GRP-R) in Cancer Therapy: Development of Bombesin-Based Peptide–Drug Conjugates. Int. J. Mol. Sci. 2023, 24, 3400. [Google Scholar] [CrossRef] [PubMed]
- Baranyai, Z.; Biri-Kovács, B.; Krátký, M.; Szeder, B.; Debreczeni, M.L.; Budai, J.; Kovács, B.; Horváth, L.; Pári, E.; Németh, Z.; et al. Cellular Internalization and Inhibition Capacity of New Anti-Glioma Peptide Conjugates: Physicochemical Characterization and Evaluation on Various Monolayer- and 3D-Spheroid-Based in Vitro Platforms. J. Med. Chem. 2021, 64, 2982–3005. [Google Scholar] [CrossRef] [PubMed]
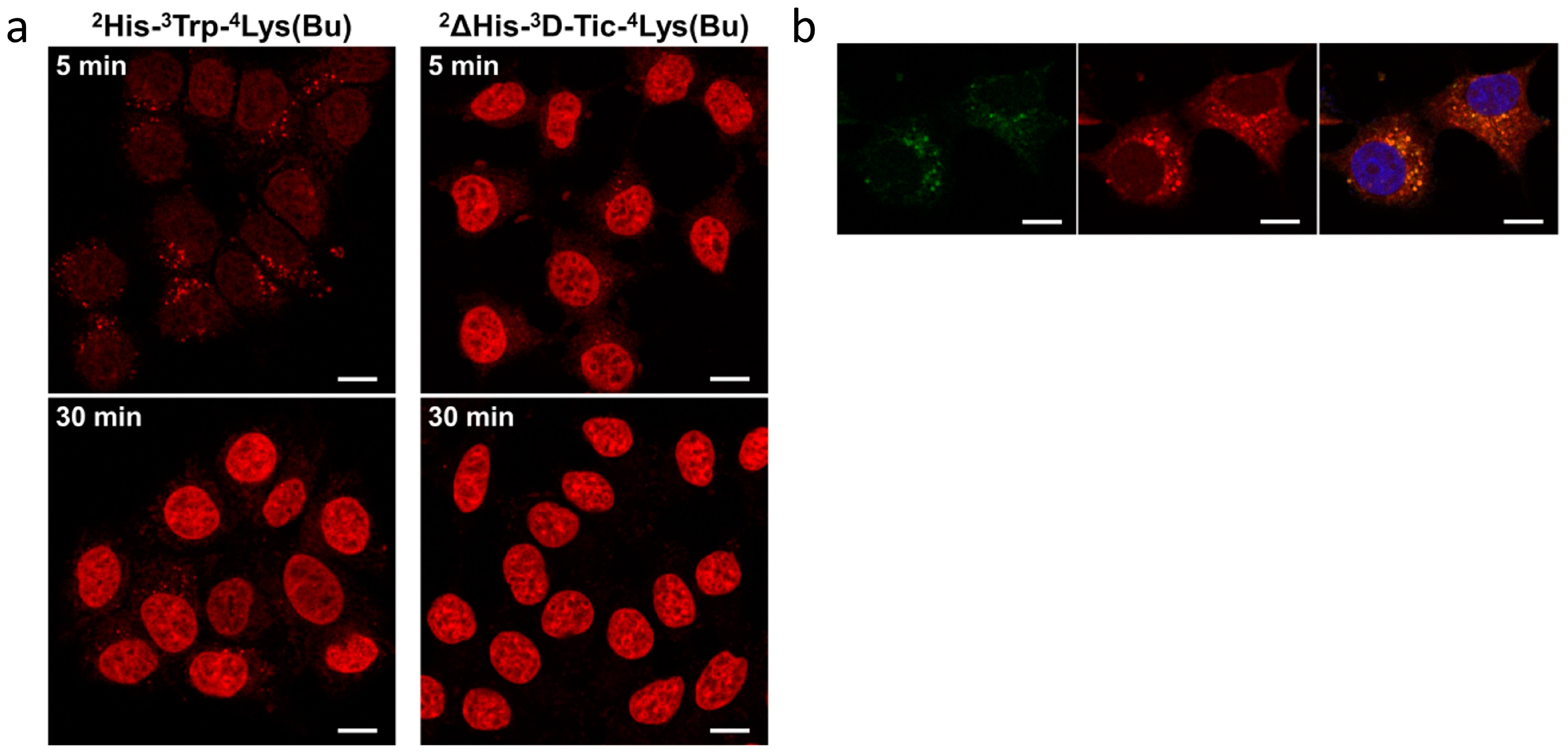
- Manea, M.; Leurs, U.; Orbán, E.; Baranyai, Z.; Öhlschläger, P.; Marquardt, A.; Schulcz, Á.; Tejeda, M.; Kapuvári, B.; Tóvári, J.; et al. Enhanced Enzymatic Stability and Antitumor Activity of Daunorubicin-GnRH-III Bioconjugates Modified in Position 4. Bioconjug. Chem. 2011, 22, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Biri-Kovács, B.; Szeder, B.; Buday, L.; Gardi, J.; Szabó, Z.; Halmos, G.; Mező, G. Enhanced In Vitro Antitumor Activity of GnRH-III-Daunorubicin Bioconjugates Influenced by Sequence Modification. Pharmaceutics 2018, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Benya, R.V. International Union of Pharmacology. LXVIII. Mammalian Bombesin Receptors: Nomenclature, Distribution, Pharmacology, Signaling, and Functions in Normal and Disease States. Pharmacol. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Álvarez, I.; Moreno, P.; Mantey, S.A.; Nakamura, T.; Nuche-Berenguer, B.; Moody, T.W.; Coy, D.H.; Jensen, R.T. Insights into Bombesin Receptors and Ligands: Highlighting Recent Advances. Peptides 2015, 72, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Wenger, S.; Schmuckli-Maurer, J.; Schaer, J.-C.; Gugger, M. Bombesin Receptor Subtypes in Human Cancers: Detection with the Universal Radioligand (125)I-[D-TYR(6), Beta-ALA(11), PHE(13), NLE(14)] Bombesin(6-14). Clin. Cancer Res. 2002, 8, 1139–1146. [Google Scholar]
- Majumdar, S.; Siahaan, T.J. Peptide-mediated Targeted Drug Delivery. Med. Res. Rev. 2012, 32, 637–658. [Google Scholar] [CrossRef]
- Preston, S.R.; Miller, G.V.; Primrose, J.N. Bombesin-like Peptides and Cancer. Crit. Rev. Oncol. Hematol. 1996, 23, 225–238. [Google Scholar] [CrossRef]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin Receptor Antagonists May Be Preferable to Agonists for Tumor Targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef]
- Valverde, I.E.; Bauman, A.; Kluba, C.A.; Vomstein, S.; Walter, M.A.; Mindt, T.L. 1,2,3-Triazoles as Amide Bond Mimics: Triazole Scan Yields Protease-Resistant Peptidomimetics for Tumor Targeting. Angew. Chem. Int. Ed. 2013, 52, 8957–8960. [Google Scholar] [CrossRef]
- Accardo, A.; Galli, F.; Mansi, R.; Del Pozzo, L.; Aurilio, M.; Morisco, A.; Ringhieri, P.; Signore, A.; Morelli, G.; Aloj, L. Pre-Clinical Evaluation of Eight DOTA Coupled Gastrin-Releasing Peptide Receptor (GRP-R) Ligands for in Vivo Targeting of Receptor-Expressing Tumors. EJNMMI Res. 2016, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Lymperis, E.; Kaloudi, A.; Kanellopoulos, P.; de Jong, M.; Krenning, E.; Nock, B.; Maina, T. Comparing Gly11/DAla11-Replacement vs. the in-Situ Neprilysin-Inhibition Approach on the Tumor-Targeting Efficacy of the 111In-SB3/111In-SB4 Radiotracer Pair. Molecules 2019, 24, 1015. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Kaloudi, A.; Lymperis, E.; Giarika, A.; Kulkarni, H.R.; Klette, I.; Singh, A.; Krenning, E.P.; de Jong, M.; Maina, T.; et al. Theranostic Perspectives in Prostate Cancer with the Gastrin-Releasing Peptide Receptor Antagonist NeoBOMB1: Preclinical and First Clinical Results. J. Nucl. Med. 2017, 58, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Identification and Stabilization of a Highly Selective Gastrin-releasing Peptide Receptor Agonist. J. Pept. Sci. 2019, 25, e3224. [Google Scholar] [CrossRef]
- Nagy, A.; Armatis, P.; Cai, R.-Z.; Szepeshazi, K.; Halmos, G.; Schally, A.V. Design, Synthesis, and in Vitro Evaluation of Cytotoxic Analogs of Bombesin-like Peptides Containing Doxorubicin or Its Intensely Potent Derivative, 2-Pyrrolinodoxorubicin. Proc. Natl. Acad. Sci. USA 1997, 94, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Deutscher, S.L. Phage Peptide Display. In Molecular Imaging II; Springer: Berlin/Heidelberg, Germany, 2008; pp. 145–163. [Google Scholar] [CrossRef]
- Gautam, A.; Kapoor, P.; Chaudhary, K.; Kumar, R.; Drug Discovery Consortium, O.; Raghava, G.P.S. Tumor Homing Peptides as Molecular Probes for Cancer Therapeutics, Diagnostics and Theranostics. Curr. Med. Chem. 2014, 21, 2367–2391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, J.; Zhang, Y.; Hu, Z.; Hu, D.; Pan, Y.; Ou, S.; Liu, G.; Yin, X.; Zhao, J.; et al. Panning and Identification of a Colon Tumor Binding Peptide from a Phage Display Peptide Library. SLAS Discov. 2007, 12, 429–435. [Google Scholar] [CrossRef]
- Basu, A.; Chakrabarti, A. Hemoglobin Interacting Proteins and Implications of Spectrin Hemoglobin Interaction. J. Proteom. 2015, 128, 469–475. [Google Scholar] [CrossRef]
- Shevtsov, M.; Huile, G.; Multhoff, G. Membrane Heat Shock Protein 70: A Theranostic Target for Cancer Therapy. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160526. [Google Scholar] [CrossRef]
- Matters, G.; Harms, J. Utilizing Peptide Ligand GPCRs to Image and Treat Pancreatic Cancer. Biomedicines 2018, 6, 65. [Google Scholar] [CrossRef]
- Sanna, V.; Nurra, S.; Pala, N.; Marceddu, S.; Pathania, D.; Neamati, N.; Sechi, M. Targeted Nanoparticles for the Delivery of Novel Bioactive Molecules to Pancreatic Cancer Cells. J. Med. Chem. 2016, 59, 5209–5220. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, J.; Ji, Y.; Lu, J.; Chan, R.; Meng, H. Targeted Drug Delivery Using IRGD Peptide for Solid Cancer Treatment. Mol. Syst. Des. Eng. 2017, 2, 370–379. [Google Scholar] [CrossRef]
- Nishimoto, T.; Yamamoto, Y.; Yoshida, K.; Goto, N.; Ohnami, S.; Aoki, K. Development of Peritoneal Tumor-Targeting Vector by In Vivo Screening with a Random Peptide-Displaying Adenovirus Library. PLoS ONE 2012, 7, e45550. [Google Scholar] [CrossRef]
- Joyce, J.A.; Laakkonen, P.; Bernasconi, M.; Bergers, G.; Ruoslahti, E.; Hanahan, D. Stage-Specific Vascular Markers Revealed by Phage Display in a Mouse Model of Pancreatic Islet Tumorigenesis. Cancer Cell 2003, 4, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Dókus, L.E.; Lajkó, E.; Ranđelović, I.; Mező, D.; Schlosser, G.; Kőhidai, L.; Tóvári, J.; Mező, G. Phage Display-Based Homing Peptide-Daunomycin Conjugates for Selective Drug Targeting to PANC-1 Pancreatic Cancer. Pharmaceutics 2020, 12, 576. [Google Scholar] [CrossRef] [PubMed]
- Feni, L.; Parente, S.; Robert, C.; Gazzola, S.; Arosio, D.; Piarulli, U.; Neundorf, I. Kiss and Run: Promoting Effective and Targeted Cellular Uptake of a Drug Delivery Vehicle Composed of an Integrin-Targeting Diketopiperazine Peptidomimetic and a Cell-Penetrating Peptide. Bioconjugate Chem. 2019, 30, 2011–2022. [Google Scholar] [CrossRef] [PubMed]
- Vrettos, E.I.; Karampelas, T.; Sayyad, N.; Kougioumtzi, A.; Syed, N.; Crook, T.; Murphy, C.; Tamvakopoulos, C.; Tzakos, A.G. Development of Programmable Gemcitabine-GnRH pro-Drugs Bearing Linker Controllable “Click” Oxime Bond Tethers and Preclinical Evaluation against Prostate Cancer. Eur. J. Med. Chem. 2021, 211, 113018. [Google Scholar] [CrossRef] [PubMed]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of Site-Specific Antibody-Drug Conjugates Using Unnatural Amino Acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, H.; Kawase, T.; Nakazawa, T.; Mori, M.; Yoshida, T. Anti-Tumor Effect of Antibody Drug Conjugate ASP1235 Targeting Fms-like Tyrosine Kinase 3 with Venetoclax plus Azacitidine in an Acute Myeloid Leukemia Xenograft Mouse Model. Oncotarget 2022, 13, 1359–1368. [Google Scholar] [CrossRef]
- Snyder, J.T.; Malinao, M.-C.; Dugal-Tessier, J.; Atkinson, J.E.; Anand, B.S.; Okada, A.; Mendelsohn, B.A. Metabolism of an Oxime-Linked Antibody Drug Conjugate, AGS62P1, and Characterization of Its Identified Metabolite. Mol. Pharm. 2018, 15, 2384–2390. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja Shastri, P.; Zhu, J.; Skidmore, L.; Liang, X.; Ji, Y.; Gu, Y.; Tian, F.; Yao, S.; Xia, G. Nonclinical Development of Next-Generation Site-Specific HER2-Targeting Antibody–Drug Conjugate (ARX788) for Breast Cancer Treatment. Mol. Cancer Ther. 2020, 19, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Barok, M.; Le Joncour, V.; Martins, A.; Isola, J.; Salmikangas, M.; Laakkonen, P.; Joensuu, H. ARX788, a Novel Anti-HER2 Antibody-Drug Conjugate, Shows Anti-Tumor Effects in Preclinical Models of Trastuzumab Emtansine-Resistant HER2-Positive Breast Cancer and Gastric Cancer. Cancer Lett. 2020, 473, 156–163. [Google Scholar] [CrossRef]
- Skidmore, L.; Sakamuri, S.; Knudsen, N.A.; Hewet, A.G.; Milutinovic, S.; Barkho, W.; Biroc, S.L.; Kirtley, J.; Marsden, R.; Storey, K.; et al. ARX788, a Site-Specific Anti-HER2 Antibody–Drug Conjugate, Demonstrates Potent and Selective Activity in HER2-Low and T-DM1–Resistant Breast and Gastric Cancers. Mol. Cancer Ther. 2020, 19, 1833–1843. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, D.; Shen, W.; Xiao, Q.; Gu, Y.; O’Shaughnessy, J.; Hu, X. Phase I Trial of a Novel Anti-HER2 Antibody–Drug Conjugate, ARX788, for the Treatment of HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2022, 28, 4212–4221. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, M.-Z.; Wang, J.-F.; Zhang, Y.-Q.; Shen, A.; Yuan, X.-L.; Zhang, T.; Wei, X.-L.; Zhao, H.-Y.; Wang, D.-S.; et al. Phase 1 Multicenter, Dose-Expansion Study of ARX788 as Monotherapy in HER2-Positive Advanced Gastric and Gastroesophageal Junction Adenocarcinoma. Cell Rep. Med. 2022, 3, 100814. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ding, J.; Xiao, C.; Li, L.; Zhuang, X.; Chen, X. Versatile Preparation of Intracellular-Acidity-Sensitive Oxime-Linked Polysaccharide-Doxorubicin Conjugate for Malignancy Therapeutic. Biomaterials 2015, 54, 72–86. [Google Scholar] [CrossRef]
- Pavličková, V.; Jurášek, M.; Rimpelová, S.; Záruba, K.; Sedlák, D.; Šimková, M.; Kodr, D.; Staňková, E.; Fähnrich, J.; Rottnerová, Z.; et al. Oxime-Based 19-Nortestosterone-Pheophorbide: A Conjugate: Bimodal Controlled Release Concept for PDT. J. Mater. Chem. B 2019, 7, 5465–5477. [Google Scholar] [CrossRef]
- Tarantino, P.; Ricciuti, B.; Pradhan, S.M.; Tolaney, S.M. Optimizing the Safety of Antibody–Drug Conjugates for Patients with Solid Tumours. Nat. Rev. Clin. Oncol. 2023, 20, 558–576. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linkage | Used Functional Groups on Peptides and Drugs | Advantage | Disadvantage | Refs. |
|---|---|---|---|---|
| Ester -CO-O- | -COOH + HO- | free drug release by esterases or pH | not fully stable in circulation; no selective bond formation, low yield; O-N acyl shift | [22,23,24] |
| Hydrazone -CO-NH-N=C- | -CO-NH-NH2 + O=C< | bond formation by chemo-selective ligation; free drug release at pH < 5 (in lysosomes) | difficulties during the purification because of acid sensitivity | [25,26,27,28] |
| Self-immolative -NH-Bz-O-CO-NH- c Y-CO-O-(CH2)2-S-S-C- | -Aaa1-Aaa2-PABC-X a + NH2- -COOH + HO-(CH2)2-S-S-C- | enzyme or GSH b cleavable spacers; the free drug can be released; it increases the distance between the drug and the homing peptide | it might influence the receptor affinity of peptide–drug conjugates | [19,29,30,31,32,33,34,35] |
| Disulfide -C-S-S-C- | -SH + HS- | drug release is easy under reductive circumstances in tumor cells | rarely used for direct drug conjugation; additional self-immolative spacer is required; the asymmetric disulfide bonds are not adequately stable | [36,37,38,39] |
| Amide -C-CO-NH-C- | -COOH + H2N- | selective conjugation is not easy; in the most cases, there is no free drug release | [19,32,40,41,42,43] | |
| Thioether -C-S-C- | -Mal c + HS- or -CO-CH2-Cl + HS- | bond formation by chemo-selective ligation (by alkylation or Michael addition) | no free drug release; disulfide byproduct during the thioether bond formation; sulfide oxidation | [35] |
| Oxime -C-O-N=C- | -CO-CH2-O-NH2 + O=C< | bond formation by chemo-selective ligation with quantitative reaction; active metabolite release | no free drug release | [20,44,45,46,47] |
| Types of the Compounds | Names of the Compounds | IC50 Values (μM) | Cardiotoxicity | ||
|---|---|---|---|---|---|
| MCF-7 a | HT-29 b | HCM c | HUVEC d | ||
| drug molecules | doxorubicin (Dox) | 0.1 ± 0.0 | 0.1 ± 0.0 | +++ | +++ |
| daunorubicin (Dau) | 0.4 ± 0.1 | 0.3 ± 0.2 | +++ | +++ | |
| reference conjugate with ester bond-linked Dox | Zoptarelin Doxorubicin | 0.2 ± 0.1 | 1.9 ± 0.3 | +++ | +++ |
| GnRH-III conjugate with ester bond-linked Dox | [8Lys(Dox-O-glut)]-GnRH-III | 0.1 ± 0.1 | 2.4 ± 0.2 | +++ | +++ |
| GnRH-III conjugate with oxime bond-linked Dau | [8Lys(Dau=Aoa)]-GnRH-III | 6.5 ± 1.8 | 27.8 ± 4.2 | 0 | 0 |
| [8Lys(Dau=Aoa-GFLG)]-GnRH-III | 3.9 ± 1.2 | 22.5 ± 1.7 | + | ++ | |
| [8Lys(Dau=Aoa-LRRY)]-GnRH-III | 1.8 ± 0.5 | 28.6 ± 5.5 | + | ++ | |
| [4Lys(Ac),8Lys(Dau=Aoa)]-GnRH-III | 3.1 ± 1.7 | 7.4 ± 2.6 | + | 0 | |
| GnRH-III conjugates with two oxime bond-linked Dau | [4Lys(Dau=Aoa),8Lys(Dau=Aoa)]-GnRH-III | 2.9 ± 0.9 | 6.8 ± 1.0 | + | 0 |
| [8Lys(Dau=Aoa-K(Dau=Aoa))]-GnRH-III | 3.0 ± 0.4 | 5.6 ± 2.0 | 0 | 0 | |
| Conjugates | Cytostatic Effect (IC50: μM) on HT-29 cells | C26 a s.c. Balb/c [%] | C26 orth. Balb/c [%] | HT-29 b s.c. SCID [%] | HT-29 orth. SCID [%] | Refs. |
|---|---|---|---|---|---|---|
| [8Lys(Dau=Aoa)]-GnRH-III | 14.2 ± 3.2 | 34.5 (v) | 7.0 (w) | 44.3 (v) 41.0 (w) | n.d. | [72,81,86] |
| [8Lys(Dau=Aoa-GFLG)]-GnRH-III | 19.4 ± 3.1 27.8 ± 4.2 | 32.3 (v) | n.d. | 57.6 (v) 50.0 (w) | n.d. | [72,81,86] |
| [4Lys(Ac),8Lys(Dau=Aoa)]-GnRH-III | 7.4 ± 2.6 | n.d. | 49.3 (w) | n.d. | 29.7 (w) | [82,86] |
| [4Lys(Bu),8Lys(Dau=Aoa)]-GnRH-III | 2.2 ± 0.6 15.9 ± 1.0 | n.d. | n.d. | n.d. | 39.4 (w) 80.7 (w) | [73,82,83,87] |
| [2ΔHis, 3D-Tic, 4Lys(Bu), 8Lys(Dau=Aoa)]-GnRH-III | 3.3 ± 0.9 | n.d. | n.d. | n.d. | 87.1 (w) | [82,87] |
| L1: Dau=Aoa-LRRY- QWAVGHLNle-NH2 | L5: Dau=Aoa-LRRY- fQWAVGHStaL-NH2 | L6: Dau=Aoa-LRRY- fQWAVβAlaHStaNle-NH2 | |
|---|---|---|---|
| cytostatic effect (IC50) | 4.38 ± 0.33 μM | 2.22 ± 0.19 μM | 18.04 ± 3.01 μM |
| cellular uptake (UC50) | 22.92 μM | 12.35 μM | 16.09 μM |
| active metabolite release in lysosomal homogenate | 80% at 6 h | 33% at 6 h | 67% at 6 h |
| mouse plasma stability | 50% intact at 3 h | 84% intact at 24 h | 73% intact at 24 h |
| tumor volume inhibition | n.d. | 21.4% | 31.4% |
| tumor weight inhibition | n.d. | 16.6% | 33.1% |
| tumor DT enhancement. | n.d. | 8.5% | 11.5% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mező, G.; Gomena, J.; Ranđelović, I.; Dókus, E.L.; Kiss, K.; Pethő, L.; Schuster, S.; Vári, B.; Vári-Mező, D.; Lajkó, E.; et al. Oxime-Linked Peptide–Daunomycin Conjugates as Good Tools for Selection of Suitable Homing Devices in Targeted Tumor Therapy: An Overview. Int. J. Mol. Sci. 2024, 25, 1864. https://doi.org/10.3390/ijms25031864
Mező G, Gomena J, Ranđelović I, Dókus EL, Kiss K, Pethő L, Schuster S, Vári B, Vári-Mező D, Lajkó E, et al. Oxime-Linked Peptide–Daunomycin Conjugates as Good Tools for Selection of Suitable Homing Devices in Targeted Tumor Therapy: An Overview. International Journal of Molecular Sciences. 2024; 25(3):1864. https://doi.org/10.3390/ijms25031864
Chicago/Turabian StyleMező, Gábor, Jacopo Gomena, Ivan Ranđelović, Endre Levente Dókus, Krisztina Kiss, Lilla Pethő, Sabine Schuster, Balázs Vári, Diána Vári-Mező, Eszter Lajkó, and et al. 2024. "Oxime-Linked Peptide–Daunomycin Conjugates as Good Tools for Selection of Suitable Homing Devices in Targeted Tumor Therapy: An Overview" International Journal of Molecular Sciences 25, no. 3: 1864. https://doi.org/10.3390/ijms25031864
APA StyleMező, G., Gomena, J., Ranđelović, I., Dókus, E. L., Kiss, K., Pethő, L., Schuster, S., Vári, B., Vári-Mező, D., Lajkó, E., Polgár, L., Kőhidai, L., Tóvári, J., & Szabó, I. (2024). Oxime-Linked Peptide–Daunomycin Conjugates as Good Tools for Selection of Suitable Homing Devices in Targeted Tumor Therapy: An Overview. International Journal of Molecular Sciences, 25(3), 1864. https://doi.org/10.3390/ijms25031864