
MLe-KCNQ2: An Artificial Intelligence Model for the Prognosis of Missense KCNQ2 Gene Variants

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results
2.1. VFI Score Comparison
2.2. Model Analysis
2.3. Ensemble Model
2.4. Severity Prediction
3. Discussion
3.1. Comparison with Other Tools
3.2. Clinical Implications
3.3. Phenotypic Discrimination
3.4. Limitations
3.5. Conclusions
4. Materials and Methods
4.1. DATASET
4.2. Variant Characterization
4.3. Model Definition, Training and Optimization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bellini, G.; Miceli, F.; Soldovieri, M.V.; del Miraglia, G.E.; Coppola, G.; Taglialatela, M. KCNQ2-Related disorders. In GeneReviews; University of Washington: Seattle, WA, USA, 2010. Available online: http://www.ncbi.nlm.nih.gov/books/NBK32534/ (accessed on 21 June 2023).
- Traynelis, J.; Silk, M.; Wang, Q.; Berkovic, S.F.; Liu, L.; Ascher, D.B.; Balding, D.J.; Petrovski, S. Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome Res. 2017, 27, 1715–1729. [Google Scholar] [CrossRef]
- Knowles, J.K.; Helbig, I.; Metcalf, C.S.; Lubbers, L.S.; Isom, L.L.; Demarest, S.; Goldberg, E.M.; George, A.L.; Jr Lerche, H.; Weckhuysen, S.; et al. Precision medicine for genetic epilepsy on the horizon: Recent advances, present challenges, and suggestions for continued progress. Epilepsia 2022, 63, 2461–2475. [Google Scholar] [CrossRef]
- McInnes, G.; Sharo, A.G.; Koleske, M.L.; Brown, J.E.H.; Norstad, M.; Adhikari, A.N.; Wang, S.; Brenner, S.E.; Halpern, J.; Koenig, B.A.; et al. Opportunities and challenges for the computational interpretation of rare variation in clinically important genes. Am. J. Hum. Genet. 2021, 108, 535–548. [Google Scholar] [CrossRef]
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V.; et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A potassium channel mutation in neonatal human epilepsy. Science 1998, 279, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Ivanovic, V.; Hendrickx, R.; Van, C.R.; Hjalgrim, H.; Moller, R.S.; Gronborg, S.; Schoonjans, A.S.; Ceulemans, B.; Heavin, S.B.; et al. Extending the KCNQ2 encephalopathy spectrum: Clinical and neuroimaging findings in 17 patients. Neurology 2013, 81, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Yamagata, T.; Kubota, M.; Arai, H.; Yamashita, S.; Nakagawa, T.; Fujii, T.; Sugai, K.; Imai, K.; Uster, T.; et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 2013, 54, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Numis, A.L.; Angriman, M.; Sullivan, J.E.; Lewis, A.J.; Striano, P.; Nabbout, R.; Cilio, M.R. KCNQ2 encephalopathy: Delineation of the electroclinical phenotype and treatment response. Neurology 2014, 82, 368–370. [Google Scholar] [CrossRef]
- Pisano, T.; Numis, A.L.; Heavin, S.B.; Weckhuysen, S.; Angriman, M.; Suls, A.; Podesta, B.; Thibert, R.L.; Shapiro, K.A.; Guerrini, R.; et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 2015, 56, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Millichap, J.J.; Park, K.L.; Tsuchida, T.; Ben-Zeev, B.; Carmant, L.; Flamini, R.; Joshi, N.; Levisohn, P.M.; Marsh, E.; Nangia, S.; et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet. 2016, 2, e96. [Google Scholar] [CrossRef]
- Goto, A.; Ishii, A.; Shibata, M.; Ihara, Y.; Cooper, E.C.; Hirose, S. Characteristics of KCNQ2 variants causing either benign neonatal epilepsy or developmental and epileptic encephalopathy. Epilepsia 2019, 60, 1870–1880. [Google Scholar] [CrossRef]
- Malerba, F.; Alberini, G.; Balagura, G.; Marchese, F.; Amadori, E.; Riva, A.; Vari, M.S.; Gennaro, E.; Madia, F.; Salpietro, V.; et al. Genotype-phenotype correlations in patients with de novo KCNQ2 pathogenic variants. Neurol. Genet. 2020, 6, e528. [Google Scholar] [CrossRef]
- Dirkx, N.; Miceli, F.; Taglialatela, M.; Weckhuysen, S. The Role of Kv7.2 in Neurodevelopment: Insights and Gaps in Our Understanding. Front Physiol 2020, 11, 570588. [Google Scholar] [CrossRef]
- Millichap, J.J.; Miceli, F.; De, M.M.; Keator, C.; Joshi, N.; Tran, B.; Soldovieri, M.V.; Ambrosino, P.; Shashi, V.; Mikati, M.A.; et al. Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia 2017, 58, e10–e15. [Google Scholar] [CrossRef]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef]
- Miosge, L.A.; Field, M.A.; Sontani, Y.; Cho, V.; Johnson, S.; Palkova, A.; Balakishnan, B.; Liang, R.; Zhang, Y.; Lyon, S.; et al. Comparison of predicted and actual consequences of missense mutations. Proc. Natl. Acad. Sci. USA 2015, 112, E5189–E5198. [Google Scholar] [CrossRef]
- Niroula, A.; Vihinen, M. How good are pathogenicity predictors in detecting benign variants? PLoS Comput. Biol. 2019, 15, e1006481. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yeung WS, B.; Chiu PC, N.; Cao, D. Computational approaches for predicting variant impact: An overview from resources, principles to applications. Front Genet. 2022, 13, 981005. [Google Scholar] [CrossRef] [PubMed]
- Leong, I.U.; Stuckey, A.; Lai, D.; Skinner, J.R.; Love, D.R. Assessment of the predictive accuracy of five in silico prediction tools, alone or in combination, and two metaservers to classify long QT syndrome gene mutations. BMC Med. Genet. 2015, 16, 34. [Google Scholar] [CrossRef] [PubMed]
- Riera, C.; Padilla, N.; de la Cruz, X. The Complementarity Between Protein-Specific and General Pathogenicity Predictors for Amino Acid Substitutions. Hum. Mutat. 2016, 37, 1013–1024. [Google Scholar] [CrossRef]
- Ritter, D.M.; Horn, P.S.; Holland, K.D. In Silico Predictions of KCNQ Variant Pathogenicity in Epilepsy. Pediatr. Neurol. 2021, 118, 48–54. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Hamp, T.; Ede, J.; Schraiber, J.G.; McRae, J.; Singer-Berk, M.; Yang, Y.; Dietrich, A.S.D.; Fiziev, P.P.; Kuderna, L.F.K.; et al. The landscape of tolerated genetic variation in humans and primates. Science 2023, 380, eabn8153. [Google Scholar] [CrossRef]
- Luppino, F.; Adzhubei, I.A.; Cassa, C.A.; Toth-Petroczy, A. DeMAG predicts the effects of variants in clinically actionable genes by integrating structural and evolutionary epistatic features. Nat. Commun. 2023, 14, 2230. [Google Scholar] [CrossRef] [PubMed]
- Danzi, M.C.; Dohrn, M.F.; Fazal, S.; Beijer, D.; Rebelo, A.P.; Cintra, V.; Zuchner, S. Deep structured learning for variant prioritization in Mendelian diseases. Nat. Commun. 2023, 14, 4167. [Google Scholar] [CrossRef] [PubMed]
- Jagota, M.; Ye, C.; Albors, C.; Rastogi, R.; Koehl, A.; Ioannidis, N.; Song, Y.S. Cross-protein transfer learning substantially improves disease variant prediction. Genome Biol. 2023, 24, 182. [Google Scholar] [CrossRef] [PubMed]
- Laine, E.; Karami, Y.; Carbone, A. GEMME: A Simple and Fast Global Epistatic Model Predicting Mutational Effects. Mol. Biol. Evol. 2019, 36, 2604–2619. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lundberg, S.M.; Lee, S.I. Explaining a series of models by propagating Shapley values. Nat. Commun. 2022, 13, 4512. [Google Scholar] [CrossRef] [PubMed]
- Livesey, B.J.; Marsh, J.A. Interpreting protein variant effects with computational predictors and deep mutational scanning. Dis. Model. Mech. 2022, 15, dmm049510. [Google Scholar] [CrossRef]
- Livesey, B.J.; Marsh, J.A. Updated benchmarking of variant effect predictors using deep mutational scanning. Mol. Syst. Biol. 2023, 19, e11474. [Google Scholar] [CrossRef]
- Frazer, J.; Notin, P.; Dias, M.; Gomez, A.; Min, J.K.; Brock, K.; Gal, Y.; Marks, D.S. Disease variant prediction with deep generative models of evolutionary data. Nature 2021, 599, 91–95. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yu, G. New insights into the pathogenicity of non-synonymous variants through multi-level analysis. Sci. Rep. 2019, 9, 1667. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Roner, S.; Mai, K.; Klinkhammer, H.; Kircher, M.; Ludwig, K.U. Predicting the pathogenicity of missense variants using features derived from AlphaFold2. Bioinformatic 2023, 39, btad280. [Google Scholar] [CrossRef]
- Brandes, N.; Goldman, G.; Wang, C.H.; Ye, C.J.; Ntranos, V. Genome-wide prediction of disease variant effects with a deep protein language model. Nat. Genet. 2023, 55, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Taroni, J.N.; Grayson, P.C.; Hu, Q.; Eddy, S.; Kretzler, M.; Merkel, P.A.; Greene, C.S. MultiPLIER: A Transfer Learning Framework for Transcriptomics Reveals Systemic Features of Rare Disease. Cell Syst. 2019, 8, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom. 2013, 14 (Suppl. S3), S3. [Google Scholar] [CrossRef]
- Wu, Y.; Li, R.; Sun, S.; Weile, J.; Roth, F.P. Improved pathogenicity prediction for rare human missense variants. Am. J. Hum. Genet. 2021, 108, 1891–1906. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Gunning, A.C.; Fryer, V.; Fasham, J.; Crosby, A.H.; Ellard, S.; Baple, E.L.; Wright, C.F. Assessing performance of pathogenicity predictors using clinically relevant variant datasets. J. Med. Genet. 2021, 58, 547–555. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.M.; Riggs, E.R.; Maglott, D.R.; Lee, J.M.; Azzariti, D.R.; Niehaus, A.; Ramos, E.M.; Martin, C.L.; Landrum, M.J.; Rehm, H.L. Using ClinVar as a Resource to Support Variant Interpretation. Curr. Protoc. Hum. Genet. 2016, 89, 8. [Google Scholar] [CrossRef] [PubMed]
- Sharo, A.G.; Zou, Y.; Adhikari, A.N.; Brenner, S.E. ClinVar and HGMD genomic variant classification accuracy has improved over time, as measured by implied disease burden. Genome Med. 2023, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.G.; Azencott, C.A.; Aicheler, F.; Gieraths, U.; MacArthur, D.G.; Samocha, K.E.; Cooper, D.N.; Stenson, P.D.; Daly, M.J.; Smoller, J.W.; et al. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum. Mutat. 2015, 36, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Stead, W.W. Clinical Implications and Challenges of Artificial Intelligence and Deep Learning. JAMA 2018, 320, 1107–1108. [Google Scholar] [CrossRef]
- Nashabat, M.; Al Qahtani, X.S.; Almakdob, S.; Altwaijri, W.; Ba-Armah, D.M.; Hundallah, K.; Al, H.A.; Al, T.S.; Maddirevula, S.; Alkuraya, F.S.; et al. The landscape of early infantile epileptic encephalopathy in a consanguineous population. Seizure 2019, 69, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Aivar, P.; Fernandez-Orth, J.; Gomis-Perez, C.; Alberdi, A.; Alaimo, A.; Rodriguez, M.S.; Giraldez, T.; Miranda, P.; Areso, P.; Villarroel, A. Surface expression and subunit specific control of steady protein levels by the kv7.2 helix A-B linker. PLoS ONE 2012, 7, e47263. [Google Scholar] [CrossRef] [PubMed]
- Milh, M.; Lacoste, C.; Cacciagli, P.; Abidi, A.; Sutera-Sardo, J.; Tzelepis, I.; Colin, E.; Badens, C.; Afenjar, A.; Coeslier, A.D.; et al. Variable clinical expression in patients with mosaicism for KCNQ2 mutations. Am. J Med. Genet. A 2015, 167A, 2314–2318. [Google Scholar] [CrossRef]
- Bosselmann, C.M.; Hedrich UB, S.; Muller, P.; Sonnenberg, L.; Parthasarathy, S.; Helbig, I.; Lerche, H.; Pfeifer, N. Predicting the functional effects of voltage-gated potassium channel missense variants with multi-task learning. EBioMedicine 2022, 81, 104115. [Google Scholar] [CrossRef]
- Heyne, H.O.; Baez-Nieto, D.; Iqbal, S.; Palmer, D.S.; Brunklaus, A.; May, P.; Johannesen, K.M.; Lauxmann, S.; Lemke, J.R.; Moller, R.S.; et al. Predicting functional effects of missense variants in voltage-gated sodium and calcium channels. Sci Transl. Med. 2020, 12, eaay6848. [Google Scholar] [CrossRef]
- Brunklaus, A.; Perez-Palma, E.; Ghanty, I.; Xinge, J.; Brilstra, E.; Ceulemans, B.; Chemaly, N.; de Lange, I.; Depienne, C.; Guerrini, R.; et al. Development and Validation of a Prediction Model for Early Diagnosis of SCN1A-Related Epilepsies. Neurology 2022, 98, e1163–e1174. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Zhang, H.; Zhao, Y.; Chen, C.; Long, J.J.; Chung, W.K.; Guan, Y.; Shen, Y. MVP predicts the pathogenicity of missense variants by deep learning. Nat. Commun. 2021, 12, 510. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Singer-Berk, M.; Watts, N.A.; Phu, W.; Goodrich, J.K.; Solomonson, M.; Rehm, H.L.; MacArthur, D.G.; O’Donnell-Luria, A. Variant interpretation using population databases: Lessons from gnomAD. Hum. Mutat. 2022, 43, 1012–1030. [Google Scholar] [CrossRef] [PubMed]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Phul, S.; Kuenze, G.; Vanoye, C.G.; Sanders, C.R.; George, A.L.; Jr Meiler, J. Predicting the functional impact of KCNQ1 variants with artificial neural networks. PLoS Comput. Biol. 2022, 18, e1010038. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Zemgulyte, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef] [PubMed]
- Larrea-Sebal, A.; Benito-Vicente, A.; Fernandez-Higuero, J.A.; Jebari-Benslaiman, S.; Galicia-Garcia, U.; Uribe, K.B.; Cenarro, A.; Ostolaza, H.; Civeira, F.; Arrasate, S.; et al. MLb-LDLr: A Machine Learning Model for Predicting the Pathogenicity of LDLr Missense Variants. JACC Basic Transl. Sci. 2021, 6, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD((R))): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Fokkema, I.F.; Kroon, M.; Lopez Hernandez, J.A.; Asscheman, D.; Lugtenburg, I.; Hoogenboom, J.; den Dunnen, J.T. The LOVD3 platform: Efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. 2021, 9, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, L.; Gao, H.; Padigepati, S.R.; McRae, J.F.; Li, Y.; Kosmicki, J.A.; Fritzilas, N.; Hakenberg, J.; Dutta, A.; Shon, J.; et al. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 2018, 50, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Montgomerie, S.; Cruz, J.A.; Shrivastava, S.; Arndt, D.; Berjanskii, M.; Wishart, D.S. PROTEUS2: A web server for comprehensive protein structure prediction and structure-based annotation. Nucleic Acids Res. 2008, 36, W202–W209. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
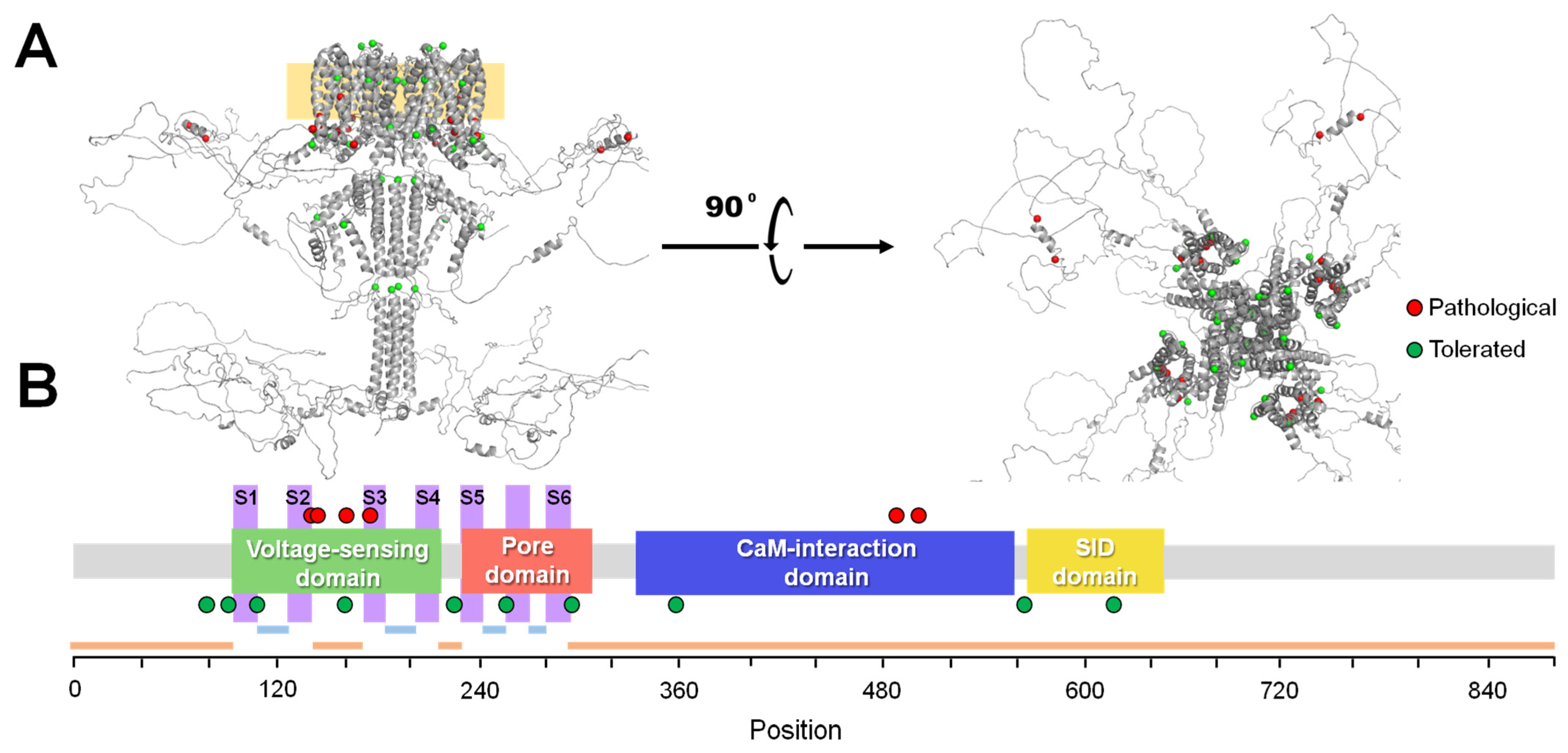{kind=link}
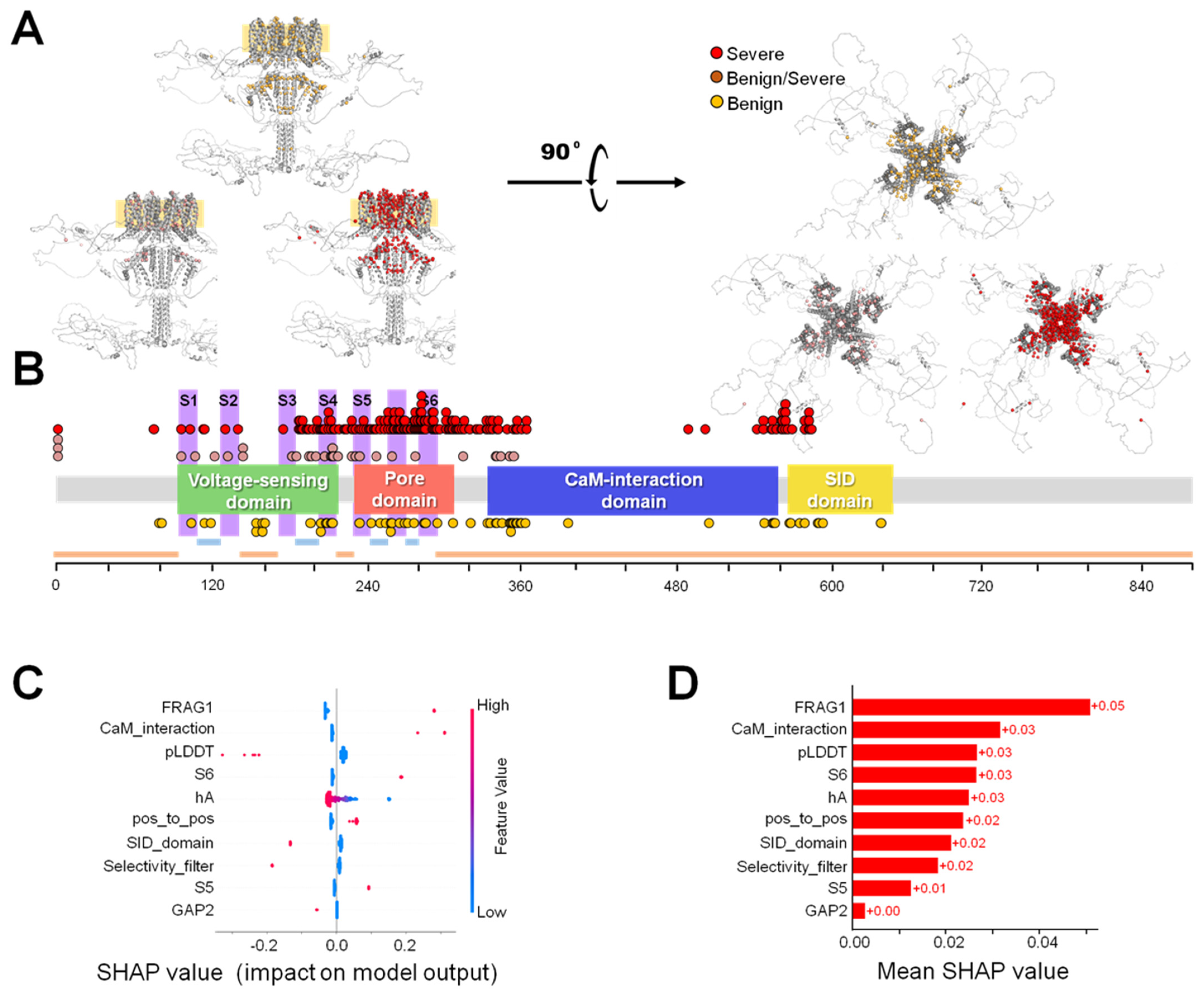{kind=link}
| Algorithm Used | AUC-ROC | Balanced Accuracy | Sensitivity | Specificity | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | Sd | Mean | Sd | Mean | Sd | Mean | Sd | ||
| VFI (σ = 2) | RF | 0.987 | 0.009 | 0.946 | 0.022 | 0.943 | 0.034 | 0.951 | 0.036 |
| VFI (σ = 3) | LR | 0.985 | 0.008 | 0.939 | 0.021 | 0.956 | 0.033 | 0.924 | 0.037 |
| VFI (σ = 4) | RF | 0.982 | 0.011 | 0.930 | 0.026 | 0.937 | 0.040 | 0.923 | 0.040 |
| pLDDT | SVM | 0.973 | 0.015 | 0.920 | 0.026 | 0.908 | 0.039 | 0.931 | 0.041 |
| AUC-ROC | Balanced Accuracy | Sensitivity | Specificity | |||||
|---|---|---|---|---|---|---|---|---|
| Mean | Sd | Mean | Sd | Mean | Sd | Mean | Sd | |
| Cross-validation | 0.993 | 0.005 | 0.961 | 0.021 | 0.966 | 0.031 | 0.956 | 0.030 |
| Test set | 0.995 | - | 0.991 | - | 0.983 | - | 1.000 | - |
| AUC-ROC | Balanced Accuracy | Sensitivity | Specificity | |||||
|---|---|---|---|---|---|---|---|---|
| Mean | Sd | Mean | Sd | Mean | Sd | Mean | Sd | |
| Cross-validation | 0.732 | 0.071 | 0.603 | 0.057 | 0.879 | 0.068 | 0.327 | 0.135 |
| Test set | 0.675 | - | 0.667 | - | 0.683 | - | 0.652 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saez-Matia, A.; Ibarluzea, M.G.; M-Alicante, S.; Muguruza-Montero, A.; Nuñez, E.; Ramis, R.; Ballesteros, O.R.; Lasa-Goicuria, D.; Fons, C.; Gallego, M.; et al. MLe-KCNQ2: An Artificial Intelligence Model for the Prognosis of Missense KCNQ2 Gene Variants. Int. J. Mol. Sci. 2024, 25, 2910. https://doi.org/10.3390/ijms25052910
Saez-Matia A, Ibarluzea MG, M-Alicante S, Muguruza-Montero A, Nuñez E, Ramis R, Ballesteros OR, Lasa-Goicuria D, Fons C, Gallego M, et al. MLe-KCNQ2: An Artificial Intelligence Model for the Prognosis of Missense KCNQ2 Gene Variants. International Journal of Molecular Sciences. 2024; 25(5):2910. https://doi.org/10.3390/ijms25052910
Chicago/Turabian StyleSaez-Matia, Alba, Markel G. Ibarluzea, Sara M-Alicante, Arantza Muguruza-Montero, Eider Nuñez, Rafael Ramis, Oscar R. Ballesteros, Diego Lasa-Goicuria, Carmen Fons, Mónica Gallego, and et al. 2024. "MLe-KCNQ2: An Artificial Intelligence Model for the Prognosis of Missense KCNQ2 Gene Variants" International Journal of Molecular Sciences 25, no. 5: 2910. https://doi.org/10.3390/ijms25052910
APA StyleSaez-Matia, A., Ibarluzea, M. G., M-Alicante, S., Muguruza-Montero, A., Nuñez, E., Ramis, R., Ballesteros, O. R., Lasa-Goicuria, D., Fons, C., Gallego, M., Casis, O., Leonardo, A., Bergara, A., & Villarroel, A. (2024). MLe-KCNQ2: An Artificial Intelligence Model for the Prognosis of Missense KCNQ2 Gene Variants. International Journal of Molecular Sciences, 25(5), 2910. https://doi.org/10.3390/ijms25052910