


Recent Advances in the Management of Diabetic Kidney Disease: Slowing Progression


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
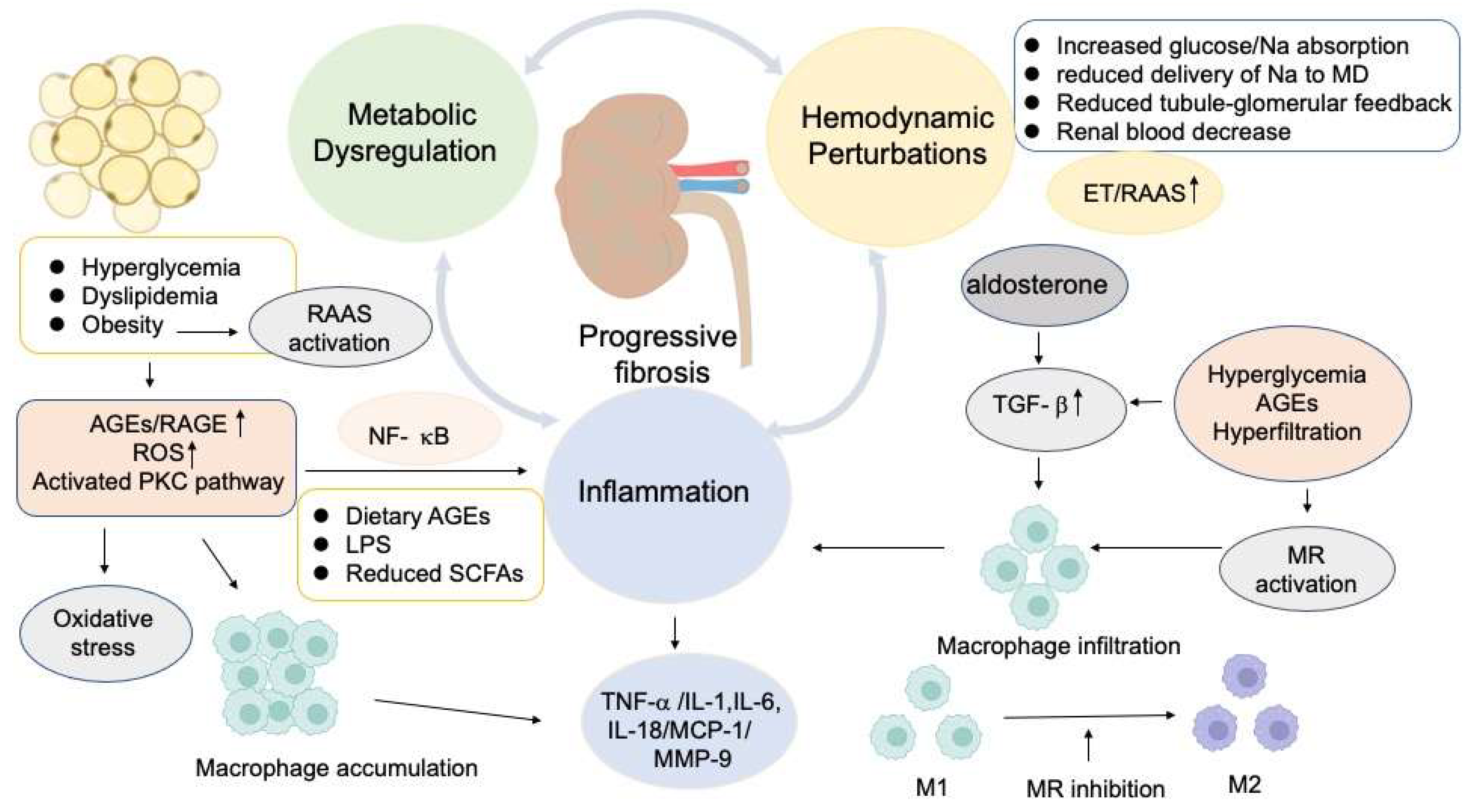
2. Molecular Mechanisms of Kidney Damage in Diabetes
2.1. Glomerular Hemodynamic Perturbations in the Pathogenesis of DKD
The Activation of Renin-Angiotensin-Aldosterone System (RAAS) in DKD
2.2. Inflammatory and Fibrotic Factors Involving in DKD
2.3. The Significance of Metabolic Factors in the Progression of DKD
2.4. The Impact of Dietary AGEs and Gut Microbiome Variation on the Progression of DKD
2.5. Genetic Predisposition and Epigenetic Modifications in the Progression of DKD
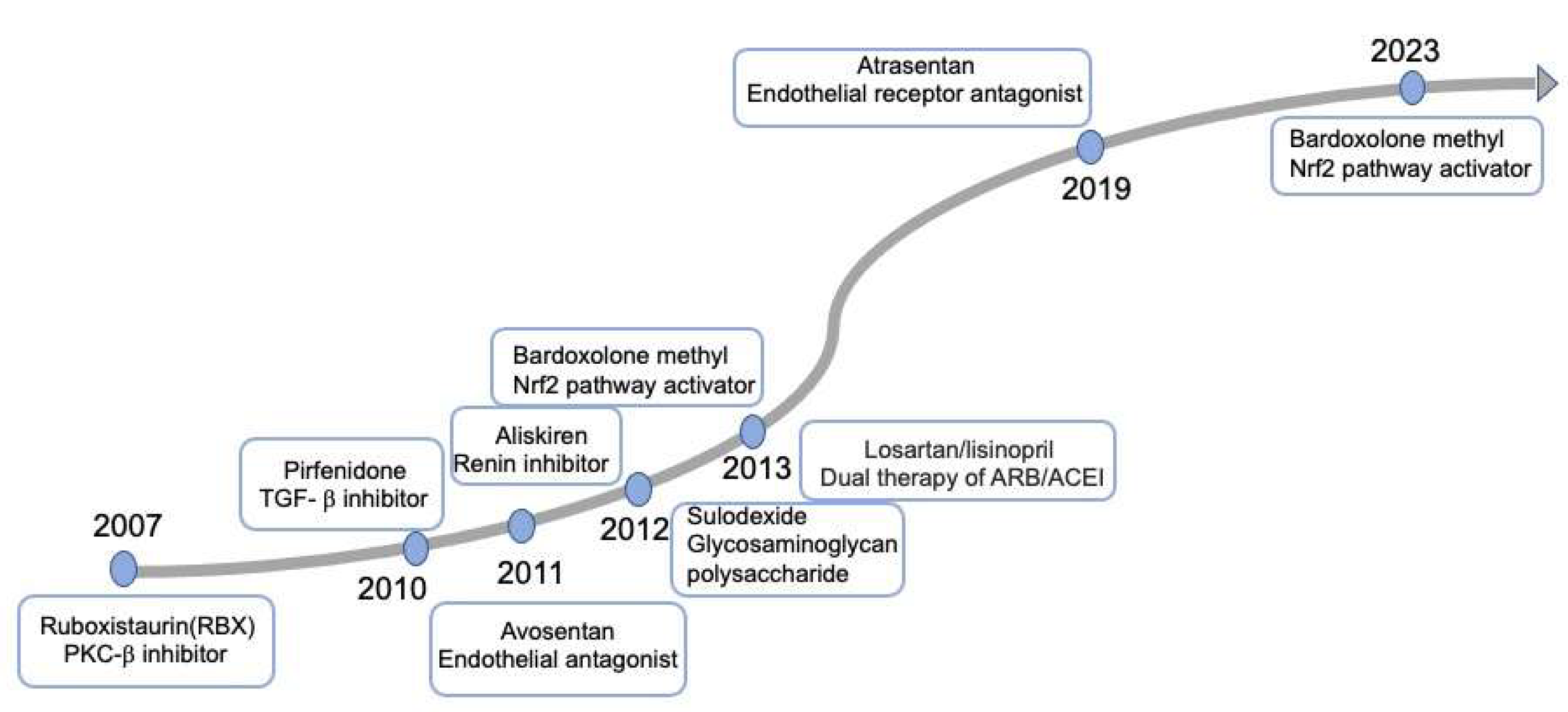
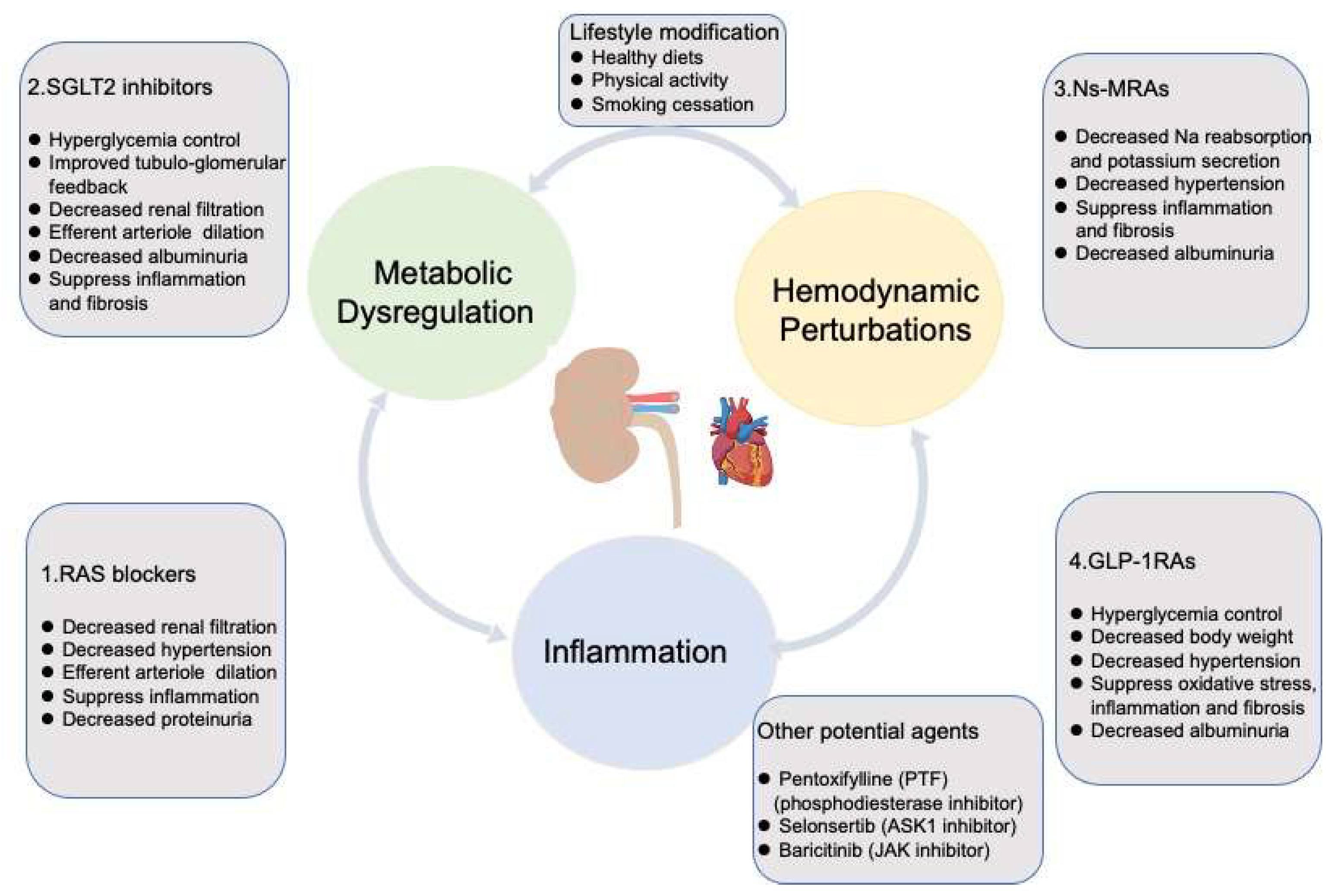
3. Targeting Mechanisms and Recent Advances in the Therapy of DKD
3.1. Renin-Angiotensin/Aldosterone System Blockades
3.2. Sodium-Glucose Cotransporter 2 Inhibitors
3.3. Nonsteroidal Mineralocorticoid Receptors Antagonists
3.4. Glucagon-like Peptide 1 Receptor Agonists
3.5. Other Agents Exhibiting Potential Effectiveness on DKD
3.6. Lifestyle Affecting the Progression of DKD
4. The Value of Multidisciplinary Treatment and Drug Combination Therapy in Clinical Application
5. Biomarkers and Future Therapeutic Targets for DKD
5.1. Molecule and Molecular Pathways as Biomarkers
5.2. Future Treatment for DKD
6. Limitations of the Study
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Nephrol. Dial. Transplant. 2019, 34, 1803–1805. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Sher, E.K.; Dzidic-Krivic, A.; Karahmet, A.; Beca-Zeco, M.; Farhat, E.K.; Softic, A.; Sher, F. Novel therapeutical approaches based on neurobiological and genetic strategies for diabetic polyneuropathy—A review. Diabetes Metab. Syndr. 2023, 17, 102901. [Google Scholar] [CrossRef] [PubMed]
- Kottgen, A.; Hwang, S.J.; Larson, M.G.; Van Eyk, J.E.; Fu, Q.; Benjamin, E.J.; Dehghan, A.; Glazer, N.L.; Kao, W.H.; Harris, T.B.; et al. Uromodulin levels associate with a common UMOD variant and risk for incident CKD. J. Am. Soc. Nephrol. 2010, 21, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R. Not all neuropathy in diabetes is of diabetic etiology: Differential diagnosis of diabetic neuropathy. Curr. Diabetes Rep. 2009, 9, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Kiernan, M.C. Uremic neuropathy: Clinical features and new pathophysiological insights. Muscle Nerve 2007, 35, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Phoon, R.K.; Pussell, B.A.; Charlesworth, J.A.; Bostock, H.; Kiernan, M.C. Altered motor nerve excitability in end-stage kidney disease. Brain 2005, 128, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Orlov, S.; Cherney, D.Z.; Pop-Busui, R.; Lovblom, L.E.; Ficociello, L.H.; Smiles, A.M.; Warram, J.H.; Krolewski, A.S.; Perkins, B.A. Cardiac autonomic neuropathy and early progressive renal decline in patients with nonmacroalbuminuric type 1 diabetes. Clin. J. Am. Soc. Nephrol. 2015, 10, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Collaboration, N.C.D.R.F. Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- Liyanage, T.; Ninomiya, T.; Jha, V.; Neal, B.; Patrice, H.M.; Okpechi, I.; Zhao, M.H.; Lv, J.; Garg, A.X.; Knight, J.; et al. Worldwide access to treatment for end-stage kidney disease: A systematic review. Lancet 2015, 385, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Agarwal, R.; Alpers, C.E.; Bakris, G.L.; Brosius, F.C.; Kolkhof, P.; Uribarri, J. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. 2022, 102, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes Diabetes Work Group. KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2022, 102, S1–S127. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Du, X.; Edelstein, D.; Obici, S.; Higham, N.; Zou, M.H.; Brownlee, M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J. Clin. Investig. 2006, 116, 1071–1080. [Google Scholar] [CrossRef]
- Paneni, F.; Beckman, J.A.; Creager, M.A.; Cosentino, F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Eur. Heart J. 2013, 34, 2436–2443. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.J. Diabetes mellitus and macrovascular disease: Mechanisms and mediators. Am. J. Med. 2007, 120, S12–S17. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.K.; Hemmelgarn, B.; Lloyd, A.; James, M.T.; Manns, B.J.; Klarenbach, S.; Tonelli, M.; Alberta Kidney Disease, N. Associations among estimated glomerular filtration rate, proteinuria, and adverse cardiovascular outcomes. Clin. J. Am. Soc. Nephrol. 2011, 6, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M.; Alberta Kidney Disease, N. Cause of Death in Patients with Reduced Kidney Function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Chang, Y.H.; Yang, S.Y.; Wu, K.D.; Chu, T.S. Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med. Assoc. 2018, 117, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Hunsicker, L.G.; Bain, R.P.; Rohde, R.D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N. Engl. J. Med. 1993, 329, 1456–1462. [Google Scholar] [CrossRef]
- Vulov, V. Infusion treatment and parenteral feeding of the newborn. Akush Ginekol 1977, 16, 387–393. [Google Scholar]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S.; et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef]
- Ma, Y.; Lin, C.; Cai, X.; Hu, S.; Zhu, X.; Lv, F.; Yang, W.; Ji, L. Baseline eGFR, albuminuria and renal outcomes in patients with SGLT2 inhibitor treatment: An updated meta-analysis. Acta Diabetol. 2023, 60, 435–445. [Google Scholar] [CrossRef]
- Kintscher, U.; Bakris, G.L.; Kolkhof, P. Novel non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Br. J. Pharmacol. 2022, 179, 3220–3234. [Google Scholar] [CrossRef]
- American Diabetes, A. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2021. Diabetes Care 2021, 44, S111–S124. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [PubMed]
- Shaman, A.M.; Bain, S.C.; Bakris, G.L.; Buse, J.B.; Idorn, T.; Mahaffey, K.W.; Mann, J.F.E.; Nauck, M.A.; Rasmussen, S.; Rossing, P.; et al. Effect of the Glucagon-Like Peptide-1 Receptor Agonists Semaglutide and Liraglutide on Kidney Outcomes in Patients with Type 2 Diabetes: Pooled Analysis of SUSTAIN 6 and LEADER. Circulation 2022, 145, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Naaman, S.C.; Bakris, G.L. Diabetic Nephropathy: Update on Pillars of Therapy Slowing Progression. Diabetes Care 2023, 46, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R. Back to the Future: Glomerular Hyperfiltration and the Diabetic Kidney. Diabetes 2017, 66, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Neumiller, J.J.; Johnson, E.J.; Dieter, B.; Tuttle, K.R. Sodium-Glucose Cotransporter 2 Inhibition and Diabetic Kidney Disease. Diabetes 2019, 68, 1094. [Google Scholar] [CrossRef]
- Guan, Z.; VanBeusecum, J.P.; Inscho, E.W. Endothelin and the renal microcirculation. Semin. Nephrol. 2015, 35, 145–155. [Google Scholar] [CrossRef]
- Lytvyn, Y.; Bjornstad, P.; van Raalte, D.H.; Heerspink, H.L.; Cherney, D.Z.I. The New Biology of Diabetic Kidney Disease-Mechanisms and Therapeutic Implications. Endocr. Rev. 2020, 41, 202–231. [Google Scholar] [CrossRef]
- Kohan, D.E.; Barton, M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int. 2014, 86, 896–904. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Miller, J.A.; Scholey, J.W.; Nasrallah, R.; Hebert, R.L.; Dekker, M.G.; Slorach, C.; Sochett, E.B.; Bradley, T.J. Renal hyperfiltration is a determinant of endothelial function responses to cyclooxygenase 2 inhibition in type 1 diabetes. Diabetes Care 2010, 33, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Premaratne, E.; Verma, S.; Ekinci, E.I.; Theverkalam, G.; Jerums, G.; MacIsaac, R.J. The impact of hyperfiltration on the diabetic kidney. Diabetes Metab. 2015, 41, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Sochett, E.B.; Cherney, D.Z.; Curtis, J.R.; Dekker, M.G.; Scholey, J.W.; Miller, J.A. Impact of renin angiotensin system modulation on the hyperfiltration state in type 1 diabetes. J. Am. Soc. Nephrol. 2006, 17, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Goodfriend, T.L.; Elliott, M.E.; Catt, K.J. Angiotensin receptors and their antagonists. N. Engl. J. Med. 1996, 334, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.M.; Wang, Z.Q.; Siragy, H.M. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 2000, 35, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Blass, G.; Palygin, O.; Levchenko, V.; Pavlov, T.S.; Grzybowski, M.N.; Winsor, K.; Shuyskiy, L.S.; Geurts, A.M.; Cowley, A.W., Jr.; et al. A NOX4/TRPC6 Pathway in Podocyte Calcium Regulation and Renal Damage in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Lorenzo, O.; Suzuki, Y.; Ruperez, M.; Egido, J. Proinflammatory actions of angiotensins. Curr. Opin. Nephrol. Hypertens. 2001, 10, 321–329. [Google Scholar] [CrossRef]
- Tesch, G.H. Macrophages and diabetic nephropathy. Semin. Nephrol. 2010, 30, 290–301. [Google Scholar] [CrossRef]
- Ritz, E.; Tomaschitz, A. Aldosterone, a vasculotoxic agent--novel functions for an old hormone. Nephrol. Dial. Transplant. 2009, 24, 2302–2305. [Google Scholar] [CrossRef]
- Tang, S.C.; Chan, L.Y.; Leung, J.C.; Cheng, A.S.; Chan, K.W.; Lan, H.Y.; Lai, K.N. Bradykinin and high glucose promote renal tubular inflammation. Nephrol. Dial. Transplant. 2010, 25, 698–710. [Google Scholar] [CrossRef]
- Tang, S.C.W.; Yiu, W.H. Innate immunity in diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef]
- Han, Q.; Zhu, H.; Chen, X.; Liu, Z. Non-genetic mechanisms of diabetic nephropathy. Front. Med. 2017, 11, 319–332. [Google Scholar] [CrossRef]
- Caamano, J.; Hunter, C.A. NF-kappaB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Pavkov, M.E.; Skupien, J.; Smiles, A.; Md Dom, Z.I.; Wilson, J.M.; Park, J.; Nair, V.; Schlafly, A.; Saulnier, P.J.; et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 2019, 25, 805–813. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Johnson, E.J.; Tuttle, K.R. Inflammatory Mechanisms as New Biomarkers and Therapeutic Targets for Diabetic Kidney Disease. Adv. Chronic Kidney Dis. 2018, 25, 181–191. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Interleukin-18 and diabetic nephropathy: A review. J. Cell Physiol. 2019, 234, 5674–5682. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Zhou, H.; Setia, O.; Liu, B.; Kanasaki, K.; Koya, D.; Dardik, A.; Fernandez-Hernando, C.; Goodwin, J. Loss of endothelial glucocorticoid receptor accelerates diabetic nephropathy. Nat. Commun. 2021, 12, 2368. [Google Scholar] [CrossRef] [PubMed]
- Pichler, R.; Afkarian, M.; Dieter, B.P.; Tuttle, K.R. Immunity and inflammation in diabetic kidney disease: Translating mechanisms to biomarkers and treatment targets. Am. J. Physiol. Renal Physiol. 2017, 312, F716–F731. [Google Scholar] [CrossRef] [PubMed]
- Schrauben, S.J.; Shou, H.; Zhang, X.; Anderson, A.H.; Bonventre, J.V.; Chen, J.; Coca, S.; Furth, S.L.; Greenberg, J.H.; Gutierrez, O.M.; et al. Association of Multiple Plasma Biomarker Concentrations with Progression of Prevalent Diabetic Kidney Disease: Findings from the Chronic Renal Insufficiency Cohort (CRIC) Study. J. Am. Soc. Nephrol. 2021, 32, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, H.; Liu, F.; Ma, Q. Up-regulation of matrix metalloproteinases-9 in the kidneys of diabetic rats and the association with neutrophil gelatinase-associated lipocalin. BMC Nephrol. 2021, 22, 211. [Google Scholar] [CrossRef]
- Yue, Y.; Yeh, J.N.; Chiang, J.Y.; Sung, P.H.; Chen, Y.L.; Liu, F.; Yip, H.K. Intrarenal arterial administration of human umbilical cord-derived mesenchymal stem cells effectively preserved the residual renal function of diabetic kidney disease in rat. Stem Cell Res. Ther. 2022, 13, 186. [Google Scholar] [CrossRef]
- Perez-Morales, R.E.; Del Pino, M.D.; Valdivielso, J.M.; Ortiz, A.; Mora-Fernandez, C.; Navarro-Gonzalez, J.F. Inflammation in Diabetic Kidney Disease. Nephron 2019, 143, 12–16. [Google Scholar] [CrossRef]
- Guiteras, R.; Flaquer, M.; Cruzado, J.M. Macrophage in chronic kidney disease. Clin. Kidney J. 2016, 9, 765–771. [Google Scholar] [CrossRef]
- Black, L.M.; Lever, J.M.; Agarwal, A. Renal Inflammation and Fibrosis: A Double-edged Sword. J. Histochem. Cytochem. 2019, 67, 663–681. [Google Scholar] [CrossRef]
- Qi, R.; Yang, C. Renal tubular epithelial cells: The neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018, 9, 1126. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.L.; Liu, T.T.; Lan, H.Y. TGF-Beta as a Master Regulator of Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 7881. [Google Scholar] [CrossRef]
- Yang, C.; Chen, X.C.; Li, Z.H.; Wu, H.L.; Jing, K.P.; Huang, X.R.; Ye, L.; Wei, B.; Lan, H.Y.; Liu, H.F. SMAD3 promotes autophagy dysregulation by triggering lysosome depletion in tubular epithelial cells in diabetic nephropathy. Autophagy 2021, 17, 2325–2344. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Cai, H.; Zhang, L.; Li, Z.; Zhong, F.; Ni, Z.; Cai, G.; Chen, X.M.; He, J.C.; Lee, K. Modulation of transforming growth factor-beta-induced kidney fibrosis by leucine-rich α-2 glycoprotein-1. Kidney Int. 2022, 101, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Typiak, M.; Piwkowska, A. Antiinflammatory Actions of Klotho: Implications for Therapy of Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 956. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Mukoyama, M.; Yanagita, M.; Yokoi, H. CTGF in kidney fibrosis and glomerulonephritis. Inflamm. Regen. 2018, 38, 14. [Google Scholar] [CrossRef]
- Brown, N.J.; Vaughan, D.E.; Fogo, A.B. The renin-angiotensin-aldosterone system and fibrinolysis in progressive renal disease. Semin. Nephrol. 2002, 22, 399–406. [Google Scholar] [CrossRef]
- Zhao, L.; Zou, Y.; Liu, F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Klemis, V.; Ghura, H.; Federico, G.; Wurfel, C.; Bentmann, A.; Gretz, N.; Miyazaki, T.; Grone, H.J.; Nakchbandi, I.A. Circulating fibronectin contributes to mesangial expansion in a murine model of type 1 diabetes. Kidney Int. 2017, 91, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Li, H.Y.; Yang, Q.; Chen, G.; Lin, S.; Liao, C.; Zhou, T. Administration of mesenchymal stem cells in diabetic kidney disease: A systematic review and meta-analysis. Stem Cell Res. Ther. 2021, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.C.; Tang, S.Q.; Liu, Y.T.; Li, A.M.; Zhan, M.; Yang, M.; Song, N.; Zhang, W.; Wu, X.Q.; Peng, C.H.; et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 2021, 12, 925. [Google Scholar] [CrossRef]
- Yang, Y.L.; Hu, F.; Xue, M.; Jia, Y.J.; Zheng, Z.J.; Li, Y.; Xue, Y.M. Early growth response protein-1 upregulates long noncoding RNA Arid2-IR to promote extracellular matrix production in diabetic kidney disease. Am. J. Physiol. Cell Physiol. 2019, 316, C340–C352. [Google Scholar] [CrossRef] [PubMed]
- Liles, J.T.; Corkey, B.K.; Notte, G.T.; Budas, G.R.; Lansdon, E.B.; Hinojosa-Kirschenbaum, F.; Badal, S.S.; Lee, M.; Schultz, B.E.; Wise, S.; et al. ASK1 contributes to fibrosis and dysfunction in models of kidney disease. J. Clin. Investig. 2018, 128, 4485–4500. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Pergola, P.E.; Chen, F.; Kirby, B.J.; Sundy, J.S.; Patel, U.D.; GS-US-223-1015 Investigators. Effects of Selonsertib in Patients with Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2019, 30, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Berthier, C.C.; Zhang, H.; Schin, M.; Henger, A.; Nelson, R.G.; Yee, B.; Boucherot, A.; Neusser, M.A.; Cohen, C.D.; Carter-Su, C.; et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 2009, 58, 469–477. [Google Scholar] [CrossRef]
- Looker, H.C.; Lin, C.; Nair, V.; Kretzler, M.; Mauer, M.; Najafian, B.; Nelson, R.G. Serum Level of Polyubiquitinated PTEN and Loss of Kidney Function in American Indians with Type 2 Diabetes. Am. J. Kidney Dis. 2022, 79, 497–506. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Q.; Li, C.; Liang, K.; Xiang, Y.; Hsiao, H.; Nguyen, T.K.; Park, P.K.; Egranov, S.D.; Ambati, C.R.; et al. PTEN-induced partial epithelial-mesenchymal transition drives diabetic kidney disease. J. Clin. Investig. 2019, 129, 1129–1151. [Google Scholar] [CrossRef]
- Lee, E.; Choi, J.; Lee, H.S. Palmitate induces mitochondrial superoxide generation and activates AMPK in podocytes. J. Cell Physiol. 2017, 232, 3209–3217. [Google Scholar] [CrossRef]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Q.; Zhang, D.D.; Wang, Y.N.; Tan, Y.Q.; Yu, X.Y.; Zhao, Y.Y. AGE/RAGE in diabetic kidney disease and ageing kidney. Free Radic. Biol. Med. 2021, 171, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Wada, T. Revisiting inflammation in diabetic nephropathy: The role of the Nlrp3 inflammasome in glomerular resident cells. Kidney Int. 2015, 87, 12–14. [Google Scholar] [CrossRef]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S.I. RAGE-Aptamer Blocks the Development and Progression of Experimental Diabetic Nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jourde-Chiche, N. Endothelial Toxicity of High Glucose and its by-Products in Diabetic Kidney Disease. Toxins 2019, 11, 578. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Yang, Z.; Zhang, C.; Shi, Y.; Han, W.; Song, S.; Mu, L.; Du, C.; Shi, Y. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism 2021, 118, 154748. [Google Scholar] [CrossRef]
- Hojs, R.; Ekart, R.; Bevc, S.; Hojs, N. Markers of Inflammation and Oxidative Stress in the Development and Progression of Renal Disease in Diabetic Patients. Nephron 2016, 133, 159–162. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, L.J.; Teichberg, S.; Fuh, H.; Li, Y.M.; Steffes, M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc. Natl. Acad. Sci. USA 1994, 91, 11704–11708. [Google Scholar] [CrossRef]
- Garay-Sevilla, M.E.; Beeri, M.S.; de la Maza, M.P.; Rojas, A.; Salazar-Villanea, S.; Uribarri, J. The potential role of dietary advanced glycation endproducts in the development of chronic non-infectious diseases: A narrative review. Nutr. Res. Rev. 2020, 33, 298–311. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.M.; Clarke, R.E.; de Pasquale, C.; Thallas-Bonke, V.; Nguyen, T.V.; Penfold, S.A.; Harcourt, B.E.; Sourris, K.C.; Lindblom, R.S.; et al. Processed foods drive intestinal barrier permeability and microvascular diseases. Sci. Adv. 2021, 7, eabe4841. [Google Scholar] [CrossRef]
- Lyte, M.; Bailey, M.T. Neuroendocrine-bacterial interactions in a neurotoxin-induced model of trauma. J. Surg. Res. 1997, 70, 195–201. [Google Scholar] [CrossRef]
- Linh, H.T.; Iwata, Y.; Senda, Y.; Sakai-Takemori, Y.; Nakade, Y.; Oshima, M.; Nakagawa-Yoneda, S.; Ogura, H.; Sato, K.; Minami, T.; et al. Intestinal Bacterial Translocation Contributes to Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Zaky, A.; Glastras, S.J.; Wong, M.Y.W.; Pollock, C.A.; Saad, S. The Role of the Gut Microbiome in Diabetes and Obesity-Related Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9641. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.D.; Leaphart, C.; Levy, R.; Prince, J.; Billiar, T.R.; Watkins, S.; Li, J.; Cetin, S.; Ford, H.; Schreiber, A.; et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J. Immunol. 2006, 176, 3070–3079. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef]
- Ma, J.; Chadban, S.J.; Zhao, C.Y.; Chen, X.; Kwan, T.; Panchapakesan, U.; Pollock, C.A.; Wu, H. TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS ONE 2014, 9, e97985. [Google Scholar] [CrossRef]
- Li, Y.J.; Chen, X.; Kwan, T.K.; Loh, Y.W.; Singer, J.; Liu, Y.; Ma, J.; Tan, J.; Macia, L.; Mackay, C.R.; et al. Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid-Mediated Activation of G Protein-Coupled Receptors GPR43 and GPR109A. J. Am. Soc. Nephrol. 2020, 31, 1267–1281. [Google Scholar] [CrossRef]
- Cole, J.B.; Florez, J.C. Genetics of diabetes mellitus and diabetes complications. Nat. Rev. Nephrol. 2020, 16, 377–390. [Google Scholar] [CrossRef]
- Sandholm, N.; Van Zuydam, N.; Ahlqvist, E.; Juliusdottir, T.; Deshmukh, H.A.; Rayner, N.W.; Di Camillo, B.; Forsblom, C.; Fadista, J.; Ziemek, D.; et al. The Genetic Landscape of Renal Complications in Type 1 Diabetes. J. Am. Soc. Nephrol. 2017, 28, 557–574. [Google Scholar] [CrossRef]
- van Zuydam, N.R.; Ahlqvist, E.; Sandholm, N.; Deshmukh, H.; Rayner, N.W.; Abdalla, M.; Ladenvall, C.; Ziemek, D.; Fauman, E.; Robertson, N.R.; et al. A Genome-Wide Association Study of Diabetic Kidney Disease in Subjects with Type 2 Diabetes. Diabetes 2018, 67, 1414–1427. [Google Scholar] [CrossRef]
- Iyengar, S.K.; Abboud, H.E.; Goddard, K.A.; Saad, M.F.; Adler, S.G.; Arar, N.H.; Bowden, D.W.; Duggirala, R.; Elston, R.C.; Hanson, R.L.; et al. Genome-wide scans for diabetic nephropathy and albuminuria in multiethnic populations: The family investigation of nephropathy and diabetes (FIND). Diabetes 2007, 56, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.; Hohenadel, D.; Brinkkoetter, P.; Peters, V.; Rind, N.; Fischer, C.; Rychlik, I.; Cerna, M.; Romzova, M.; de Heer, E.; et al. Carnosine as a protective factor in diabetic nephropathy: Association with a leucine repeat of the carnosinase gene CNDP1. Diabetes 2005, 54, 2320–2327. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Yang, Z.; Patel, S.; Chen, H.; Gibbs, D.; Yang, X.; Hau, V.S.; Kaminoh, Y.; Harmon, J.; Pearson, E.; et al. Promoter polymorphism of the erythropoietin gene in severe diabetic eye and kidney complications. Proc. Natl. Acad. Sci. USA 2008, 105, 6998–7003. [Google Scholar] [CrossRef] [PubMed]
- Sandholm, N.; Salem, R.M.; McKnight, A.J.; Brennan, E.P.; Forsblom, C.; Isakova, T.; McKay, G.J.; Williams, W.W.; Sadlier, D.M.; Makinen, V.P.; et al. New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet. 2012, 8, e1002921. [Google Scholar] [CrossRef] [PubMed]
- Salem, R.M.; Todd, J.N.; Sandholm, N.; Cole, J.B.; Chen, W.M.; Andrews, D.; Pezzolesi, M.G.; McKeigue, P.M.; Hiraki, L.T.; Qiu, C.; et al. Genome-Wide Association Study of Diabetic Kidney Disease Highlights Biology Involved in Glomerular Basement Membrane Collagen. J. Am. Soc. Nephrol. 2019, 30, 2000–2016. [Google Scholar] [CrossRef]
- Iyengar, S.K.; Sedor, J.R.; Freedman, B.I.; Kao, W.H.; Kretzler, M.; Keller, B.J.; Abboud, H.E.; Adler, S.G.; Best, L.G.; Bowden, D.W.; et al. Genome-Wide Association and Trans-ethnic Meta-Analysis for Advanced Diabetic Kidney Disease: Family Investigation of Nephropathy and Diabetes (FIND). PLoS Genet. 2015, 11, e1005352. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Keaton, J.M.; Dimitrov, L.; Hicks, P.J.; Xu, J.; Palmer, N.D.; Ma, L.; Das, S.K.; Chen, Y.I.; Coresh, J.; et al. Genome-wide association study identifies novel loci for type 2 diabetes-attributed end-stage kidney disease in African Americans. Hum. Genom. 2019, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Qiu, C.; Liu, H.; Gluck, C.; Hsu, J.Y.; He, J.; Hsu, C.Y.; Sha, D.; Weir, M.R.; Isakova, T.; et al. Systematic integrated analysis of genetic and epigenetic variation in diabetic kidney disease. Proc. Natl. Acad. Sci. USA 2020, 117, 29013–29024. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, K.; Xu, K.; Chen, S.; Cao, Y.; Zhan, H. Roles of Identified Long Noncoding RNA in Diabetic Nephropathy. J. Diabetes Res. 2019, 2019, 5383010. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Goodwin, J.E.; Tripathi, P.; Kanasaki, K.; Koya, D. Interactions among Long Non-Coding RNAs and microRNAs Influence Disease Phenotype in Diabetes and Diabetic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 6027. [Google Scholar] [CrossRef]
- Lin, J.; Jiang, Z.; Liu, C.; Zhou, D.; Song, J.; Liao, Y.; Chen, J. Emerging Roles of Long Non-Coding RNAs in Renal Fibrosis. Life 2020, 10, 131. [Google Scholar] [CrossRef]
- Hu, M.; Wang, R.; Li, X.; Fan, M.; Lin, J.; Zhen, J.; Chen, L.; Lv, Z. LncRNA MALAT1 is dysregulated in diabetic nephropathy and involved in high glucose-induced podocyte injury via its interplay with beta-catenin. J. Cell Mol. Med. 2017, 21, 2732–2747. [Google Scholar] [CrossRef]
- Alvarez, M.L.; DiStefano, J.K. Functional characterization of the plasmacytoma variant translocation 1 gene (PVT1) in diabetic nephropathy. PLoS ONE 2011, 6, e18671. [Google Scholar] [CrossRef]
- Tuttle, K.R.; McGill, J.B.; Haney, D.J.; Lin, T.E.; Anderson, P.W.; Pkc-Drs, P.-D.; Groups, P.-D.S. Kidney outcomes in long-term studies of ruboxistaurin for diabetic eye disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.; Green, D.; Jamerson, K.; Ruilope, L.M.; Kuranoff, S.J.; Littke, T.; Viberti, G.; Group, A.S. Avosentan for overt diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Rossing, P.; Caramori, M.L.; Chan, J.C.N.; Heerspink, H.J.L.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A.; et al. Executive summary of the KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease: An update based on rapidly emerging new evidence. Kidney Int. 2022, 102, 990–999. [Google Scholar] [CrossRef]
- Sharma, K.; Ix, J.H.; Mathew, A.V.; Cho, M.; Pflueger, A.; Dunn, S.R.; Francos, B.; Sharma, S.; Falkner, B.; McGowan, T.A.; et al. Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Packham, D.K.; Wolfe, R.; Reutens, A.T.; Berl, T.; Heerspink, H.L.; Rohde, R.; Ivory, S.; Lewis, J.; Raz, I.; Wiegmann, T.B.; et al. Sulodexide fails to demonstrate renoprotection in overt type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.H.; Brenner, B.M.; McMurray, J.J.; de Zeeuw, D.; Haffner, S.M.; Solomon, S.D.; Chaturvedi, N.; Persson, F.; Desai, A.S.; Nicolaides, M.; et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N. Engl. J. Med. 2012, 367, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef]
- Nangaku, M.; Kanda, H.; Takama, H.; Ichikawa, T.; Hase, H.; Akizawa, T. Randomized Clinical Trial on the Effect of Bardoxolone Methyl on GFR in Diabetic Kidney Disease Patients (TSUBAKI Study). Kidney Int. Rep. 2020, 5, 879–890. [Google Scholar] [CrossRef]
- Nangaku, M.; Takama, H.; Ichikawa, T.; Mukai, K.; Kojima, M.; Suzuki, Y.; Watada, H.; Wada, T.; Ueki, K.; Narita, I.; et al. Randomized, double-blind, placebo-controlled phase 3 study of bardoxolone methyl in patients with diabetic kidney disease: Design and baseline characteristics of the AYAME study. Nephrol. Dial. Transplant. 2023, 38, 1204–1216. [Google Scholar] [CrossRef]
- Palic, B.; Brizic, I.; Sher, E.K.; Cvetkovic, I.; Dzidic-Krivic, A.; Abdelghani, H.T.M.; Sher, F. Effects of Zofenopril on Arterial Stiffness in Hypertension Patients. Mol. Biotechnol. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Desideri, G.; Grassi, D.; Croce, G.; Bocale, R.; Tiberti, S.; Evangelista, S.; Necozione, S.; Di Orio, F.; Ferri, C. Different effects of angiotensin converting enzyme inhibitors on endothelin-1 and nitric oxide balance in human vascular endothelial cells: Evidence of an oxidant-sensitive pathway. Mediat. Inflamm. 2008, 2008, 305087. [Google Scholar] [CrossRef]
- Malacco, E.; Omboni, S.; Parati, G. Blood Pressure Response to Zofenopril or Irbesartan Each Combined with Hydrochlorothiazide in High-Risk Hypertensives Uncontrolled by Monotherapy: A Randomized, Double-Blind, Controlled, Parallel Group, Noninferiority Trial. Int. J. Hypertens. 2015, 2015, 139465. [Google Scholar] [CrossRef]
- Modesti, P.A.; Omboni, S.; Taddei, S.; Ghione, S.; Portaluppi, F.; Pozzilli, P.; Volpe, M.; Arca, M.; Calabro, P.; Fulgheri, P.L.; et al. Zofenopril or irbesartan plus hydrochlorothiazide in elderly patients with isolated systolic hypertension untreated or uncontrolled by previous treatment: A double-blind, randomized study. J. Hypertens. 2016, 34, 576–587. [Google Scholar] [CrossRef]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I.; et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef]
- Fried, L.F.; Emanuele, N.; Zhang, J.H.; Brophy, M.; Conner, T.A.; Duckworth, W.; Leehey, D.J.; McCullough, P.A.; O’Connor, T.; Palevsky, P.M.; et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N. Engl. J. Med. 2013, 369, 1892–1903. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef]
- American Diabetes, A. 11. Microvascular Complications and Foot Care: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, S135–S151. [Google Scholar] [CrossRef]
- Wanner, C.; Marx, N. SGLT2 inhibitors: The future for treatment of type 2 diabetes mellitus and other chronic diseases. Diabetologia 2018, 61, 2134–2139. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Salah, H.M.; Al’Aref, S.J.; Khan, M.S.; Al-Hawwas, M.; Vallurupalli, S.; Mehta, J.L.; Mounsey, J.P.; Greene, S.J.; McGuire, D.K.; Lopes, R.D.; et al. Effect of sodium-glucose cotransporter 2 inhibitors on cardiovascular and kidney outcomes-Systematic review and meta-analysis of randomized placebo-controlled trials. Am. Heart J. 2021, 232, 10–22. [Google Scholar] [CrossRef]
- The, E.-K.C.G.; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Girerd, S.; Jaisser, F. Mineralocorticoid receptor antagonists and kidney diseases: Pathophysiological basis. Kidney Int. 2019, 96, 302–319. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.S.; Tostes, R.C.; Paradis, P.; Schiffrin, E.L. Aldosterone, Inflammation, Immune System, and Hypertension. Am. J. Hypertens. 2021, 34, 15–27. [Google Scholar] [CrossRef]
- Tesch, G.H.; Young, M.J. Mineralocorticoid Receptor Signaling as a Therapeutic Target for Renal and Cardiac Fibrosis. Front. Pharmacol. 2017, 8, 313. [Google Scholar] [CrossRef]
- Sato, A.; Hayashi, K.; Naruse, M.; Saruta, T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension 2003, 41, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Miric, G.; Dallemagne, C.; Endre, Z.; Margolin, S.; Taylor, S.M.; Brown, L. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br. J. Pharmacol. 2001, 133, 687–694. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. Corrigendum to: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 4901. [Google Scholar] [CrossRef]
- Charytan, D.M.; Himmelfarb, J.; Ikizler, T.A.; Raj, D.S.; Hsu, J.Y.; Landis, J.R.; Anderson, A.H.; Hung, A.M.; Mehrotra, R.; Sharma, S.; et al. Safety and cardiovascular efficacy of spironolactone in dialysis-dependent ESRD (SPin-D): A randomized, placebo-controlled, multiple dosage trial. Kidney Int. 2019, 95, 973–982. [Google Scholar] [CrossRef]
- Agarwal, R.; Filippatos, G.; Pitt, B.; Anker, S.D.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Gebel, M.; Ruilope, L.M.; et al. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: The FIDELITY pooled analysis. Eur. Heart J. 2022, 43, 474–484. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Agarwal, R.; Pitt, B.; Palmer, B.F.; Kovesdy, C.P.; Burgess, E.; Filippatos, G.; Malyszko, J.; Ruilope, L.M.; Rossignol, P.; Rossing, P.; et al. A comparative post hoc analysis of finerenone and spironolactone in resistant hypertension in moderate-to-advanced chronic kidney disease. Clin. Kidney J. 2023, 16, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Kashihara, N.; Shikata, K.; Nangaku, M.; Wada, T.; Okuda, Y.; Sawanobori, T. Esaxerenone (CS-3150) in Patients with Type 2 Diabetes and Microalbuminuria (ESAX-DN): Phase 3 Randomized Controlled Clinical Trial. Clin. J. Am. Soc. Nephrol. 2020, 15, 1715–1727. [Google Scholar] [CrossRef]
- Kolkhof, P.; Hartmann, E.; Freyberger, A.; Pavkovic, M.; Mathar, I.; Sandner, P.; Droebner, K.; Joseph, A.; Huser, J.; Eitner, F. Effects of Finerenone Combined with Empagliflozin in a Model of Hypertension-Induced End-Organ Damage. Am. J. Nephrol. 2021, 52, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Kristensen, S.L.; Bengtsson, O.; Bohm, M.; de Boer, R.A.; Docherty, K.F.; Inzucchi, S.E.; Katova, T.; Kober, L.; Kosiborod, M.N.; et al. Dapagliflozin in HFrEF Patients Treated with Mineralocorticoid Receptor Antagonists: An Analysis of DAPA-HF. JACC Heart Fail. 2021, 9, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.P.; Zannad, F.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Jamal, W.; Steubl, D.; Schueler, E.; et al. Interplay of Mineralocorticoid Receptor Antagonists and Empagliflozin in Heart Failure: EMPEROR-Reduced. J. Am. Coll. Cardiol. 2021, 77, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Joseph, A.; Anker, S.D.; Filippatos, G.; Rossing, P.; Ruilope, L.M.; Pitt, B.; Kolkhof, P.; Scott, C.; Lawatscheck, R.; et al. Hyperkalemia Risk with Finerenone: Results from the FIDELIO-DKD Trial. J. Am. Soc. Nephrol. 2022, 33, 225–237. [Google Scholar] [CrossRef]
- Rossing, P.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Chan, J.C.N.; Kooy, A.; McCafferty, K.; Schernthaner, G.; et al. Finerenone in Predominantly Advanced CKD and Type 2 Diabetes with or without Sodium-Glucose Cotransporter-2 Inhibitor Therapy. Kidney Int. Rep. 2022, 7, 36–45. [Google Scholar] [CrossRef] [PubMed]
- de Boer, I.H.; Khunti, K.; Sadusky, T.; Tuttle, K.R.; Neumiller, J.J.; Rhee, C.M.; Rosas, S.E.; Rossing, P.; Bakris, G. Diabetes management in chronic kidney disease: A consensus report by the American Diabetes Association (ADA) and Kidney Disease: Improving Global Outcomes (KDIGO). Diabetes Care 2022, 45, 3075–3090. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Cox, E.J.; Neumiller, J.J.; Tuttle, K.R. Incretin drugs in diabetic kidney disease: Biological mechanisms and clinical evidence. Nat. Rev. Nephrol. 2021, 17, 227–244. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes Diabetes Work Group. KDIGO 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2020, 98, S1–S115. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Lakshmanan, M.C.; Rayner, B.; Busch, R.S.; Zimmermann, A.G.; Woodward, D.B.; Botros, F.T. Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7): A multicentre, open-label, randomised trial. Lancet Diabetes Endocrinol. 2018, 6, 605–617. [Google Scholar] [CrossRef]
- Sattar, N.; Lee, M.M.Y.; Kristensen, S.L.; Branch, K.R.H.; Del Prato, S.; Khurmi, N.S.; Lam, C.S.P.; Lopes, R.D.; McMurray, J.J.V.; Pratley, R.E.; et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021, 9, 653–662. [Google Scholar] [CrossRef]
- Palmer, S.C.; Tendal, B.; Mustafa, R.A.; Vandvik, P.O.; Li, S.; Hao, Q.; Tunnicliffe, D.; Ruospo, M.; Natale, P.; Saglimbene, V.; et al. Sodium-glucose cotransporter protein-2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists for type 2 diabetes: Systematic review and network meta-analysis of randomised controlled trials. BMJ 2021, 372, m4573. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jodar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; Sattar, N.; Rosenstock, J.; Ramasundarahettige, C.; Pratley, R.; Lopes, R.D.; Lam, C.S.P.; Khurmi, N.S.; Heenan, L.; Del Prato, S.; et al. Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, H.; Henriquez, G.; Abramowitz, M.K.; Kramer, H.; Rosas, S.E.; Johns, T.; Kumar, J.; Skversky, A.; Kaskel, F.; Melamed, M.L. Abdominal Obesity, Race and Chronic Kidney Disease in Young Adults: Results from NHANES 1999–2010. PLoS ONE 2016, 11, e0153588. [Google Scholar] [CrossRef]
- Ejerblad, E.; Fored, C.M.; Lindblad, P.; Fryzek, J.; McLaughlin, J.K.; Nyren, O. Obesity and risk for chronic renal failure. J. Am. Soc. Nephrol. 2006, 17, 1695–1702. [Google Scholar] [CrossRef]
- Rossing, P.; Baeres, F.M.M.; Bakris, G.; Bosch-Traberg, H.; Gislum, M.; Gough, S.C.L.; Idorn, T.; Lawson, J.; Mahaffey, K.W.; Mann, J.F.E.; et al. The rationale, design and baseline data of FLOW, a kidney outcomes trial with once-weekly semaglutide in people with type 2 diabetes and chronic kidney disease. Nephrol. Dial. Transplant. 2023, 38, 2041–2051. [Google Scholar] [CrossRef]
- Wright, A.K.; Carr, M.J.; Kontopantelis, E.; Leelarathna, L.; Thabit, H.; Emsley, R.; Buchan, I.; Mamas, M.A.; van Staa, T.P.; Sattar, N.; et al. Primary Prevention of Cardiovascular and Heart Failure Events with SGLT2 Inhibitors, GLP-1 Receptor Agonists, and Their Combination in Type 2 Diabetes. Diabetes Care 2022, 45, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Davila-Esqueda, M.E.; Martinez-Morales, F. Pentoxifylline diminishes the oxidative damage to renal tissue induced by streptozotocin in the rat. Exp. Diabesity Res. 2004, 5, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Gonzalez, J.F.; Mora-Fernandez, C.; Muros de Fuentes, M.; Chahin, J.; Mendez, M.L.; Gallego, E.; Macia, M.; del Castillo, N.; Rivero, A.; Getino, M.A.; et al. Effect of pentoxifylline on renal function and urinary albumin excretion in patients with diabetic kidney disease: The PREDIAN trial. J. Am. Soc. Nephrol. 2015, 26, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Gonzalez, J.F.; Sanchez-Nino, M.D.; Donate-Correa, J.; Martin-Nunez, E.; Ferri, C.; Perez-Delgado, N.; Gorriz, J.L.; Martinez-Castelao, A.; Ortiz, A.; Mora-Fernandez, C. Effects of Pentoxifylline on Soluble Klotho Concentrations and Renal Tubular Cell Expression in Diabetic Kidney Disease. Diabetes Care 2018, 41, 1817–1820. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Brosius, F.C., 3rd; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: Results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transplant. 2018, 33, 1950–1959. [Google Scholar] [CrossRef] [PubMed]
- Fragoulis, G.E.; McInnes, I.B.; Siebert, S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology 2019, 58, i43–i54. [Google Scholar] [CrossRef]
- Ko, G.J.; Rhee, C.M.; Kalantar-Zadeh, K.; Joshi, S. The Effects of High-Protein Diets on Kidney Health and Longevity. J. Am. Soc. Nephrol. 2020, 31, 1667–1679. [Google Scholar] [CrossRef]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diabetes Rep. 2014, 14, 453. [Google Scholar] [CrossRef]
- Bach, K.E.; Kelly, J.T.; Palmer, S.C.; Khalesi, S.; Strippoli, G.F.M.; Campbell, K.L. Healthy Dietary Patterns and Incidence of CKD: A Meta-Analysis of Cohort Studies. Clin. J. Am. Soc. Nephrol. 2019, 14, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.B.D.D. Health effects of dietary risks in 195 countries, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2019, 393, 1958–1972. [Google Scholar] [CrossRef]
- Pandey, A.; Garg, S.; Khunger, M.; Darden, D.; Ayers, C.; Kumbhani, D.J.; Mayo, H.G.; de Lemos, J.A.; Berry, J.D. Dose-Response Relationship Between Physical Activity and Risk of Heart Failure: A Meta-Analysis. Circulation 2015, 132, 1786–1794. [Google Scholar] [CrossRef]
- Bowlby, W.; Zelnick, L.R.; Henry, C.; Himmelfarb, J.; Kahn, S.E.; Kestenbaum, B.; Robinson-Cohen, C.; Utzschneider, K.M.; de Boer, I.H. Physical activity and metabolic health in chronic kidney disease: A cross-sectional study. BMC Nephrol. 2016, 17, 187. [Google Scholar] [CrossRef]
- Kosmadakis, G.C.; John, S.G.; Clapp, E.L.; Viana, J.L.; Smith, A.C.; Bishop, N.C.; Bevington, A.; Owen, P.J.; McIntyre, C.W.; Feehally, J. Benefits of regular walking exercise in advanced pre-dialysis chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 997–1004. [Google Scholar] [CrossRef]
- Xia, J.; Wang, L.; Ma, Z.; Zhong, L.; Wang, Y.; Gao, Y.; He, L.; Su, X. Cigarette smoking and chronic kidney disease in the general population: A systematic review and meta-analysis of prospective cohort studies. Nephrol. Dial. Transplant. 2017, 32, 475–487. [Google Scholar] [CrossRef]
- Jhee, J.H.; Joo, Y.S.; Kee, Y.K.; Jung, S.Y.; Park, S.; Yoon, C.Y.; Han, S.H.; Yoo, T.H.; Kang, S.W.; Park, J.T. Secondhand Smoke and CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 515–522. [Google Scholar] [CrossRef]
- Ueki, K.; Sasako, T.; Okazaki, Y.; Kato, M.; Okahata, S.; Katsuyama, H.; Haraguchi, M.; Morita, A.; Ohashi, K.; Hara, K.; et al. Effect of an intensified multifactorial intervention on cardiovascular outcomes and mortality in type 2 diabetes (J-DOIT3): An open-label, randomised controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 951–964. [Google Scholar] [CrossRef]
- Oshima, M.; Jardine, M.J.; Agarwal, R.; Bakris, G.; Cannon, C.P.; Charytan, D.M.; de Zeeuw, D.; Edwards, R.; Greene, T.; Levin, A.; et al. Insights from CREDENCE trial indicate an acute drop in estimated glomerular filtration rate during treatment with canagliflozin with implications for clinical practice. Kidney Int. 2021, 99, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Kraus, B.J.; Weir, M.R.; Bakris, G.L.; Mattheus, M.; Cherney, D.Z.I.; Sattar, N.; Heerspink, H.J.L.; Ritter, I.; von Eynatten, M.; Zinman, B.; et al. Characterization and implications of the initial estimated glomerular filtration rate ‘dip’ upon sodium-glucose cotransporter-2 inhibition with empagliflozin in the EMPA-REG OUTCOME trial. Kidney Int. 2021, 99, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, M.; Puchades, M.J.; Garofalo, C.; Jongs, N.; D’Marco, L.; Andreucci, M.; De Nicola, L.; Gorriz, J.L.; Heerspink, H.J.L.; group, R.-S.; et al. Albuminuria-Lowering Effect of Dapagliflozin, Eplerenone, and Their Combination in Patients with Chronic Kidney Disease: A Randomized Crossover Clinical Trial. J. Am. Soc. Nephrol. 2022, 33, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Green, J.B.; Mottl, A.K.; Bakris, G.; Heerspink, H.J.L.; Mann, J.F.E.; McGill, J.B.; Nangaku, M.; Rossing, P.; Scott, C.; Gay, A.; et al. Design of the COmbinatioN effect of FInerenone anD EmpaglifloziN in participants with chronic kidney disease and type 2 diabetes using a UACR Endpoint study (CONFIDENCE). Nephrol. Dial. Transplant. 2023, 38, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.C.B.; Cheung, C.L.; Lee, A.C.H.; Lam, J.K.Y.; Wong, Y.; Shiu, S.W.M. Galectin-3 is independently associated with progression of nephropathy in type 2 diabetes mellitus. Diabetologia 2018, 61, 1212–1219. [Google Scholar] [CrossRef]
- Scurt, F.G.; Menne, J.; Brandt, S.; Bernhardt, A.; Mertens, P.R.; Haller, H.; Chatzikyrkou, C.; Committee, R.S. Systemic Inflammation Precedes Microalbuminuria in Diabetes. Kidney Int. Rep. 2019, 4, 1373–1386. [Google Scholar] [CrossRef]
- Lampropoulou, I.T.; Stangou, M.; Papagianni, A.; Didangelos, T.; Iliadis, F.; Efstratiadis, G. TNF-alpha and microalbuminuria in patients with type 2 diabetes mellitus. J. Diabetes Res. 2014, 2014, 394206. [Google Scholar] [CrossRef]
- Saulnier, P.J.; Gand, E.; Ragot, S.; Ducrocq, G.; Halimi, J.M.; Hulin-Delmotte, C.; Llaty, P.; Montaigne, D.; Rigalleau, V.; Roussel, R.; et al. Association of serum concentration of TNFR1 with all-cause mortality in patients with type 2 diabetes and chronic kidney disease: Follow-up of the SURDIAGENE Cohort. Diabetes Care 2014, 37, 1425–1431. [Google Scholar] [CrossRef]
- Nastase, M.V.; Zeng-Brouwers, J.; Beckmann, J.; Tredup, C.; Christen, U.; Radeke, H.H.; Wygrecka, M.; Schaefer, L. Biglycan, a novel trigger of Th1 and Th17 cell recruitment into the kidney. Matrix Biol. 2018, 68–69, 293–317. [Google Scholar] [CrossRef]
- Tofte, N.; Lindhardt, M.; Adamova, K.; Bakker, S.J.L.; Beige, J.; Beulens, J.W.J.; Birkenfeld, A.L.; Currie, G.; Delles, C.; Dimos, I.; et al. Early detection of diabetic kidney disease by urinary proteomics and subsequent intervention with spironolactone to delay progression (PRIORITY): A prospective observational study and embedded randomised placebo-controlled trial. Lancet Diabetes Endocrinol. 2020, 8, 301–312. [Google Scholar] [CrossRef]
- Zhou, L.J.; Yang, D.W.; Ou, L.N.; Guo, X.R.; Wu, B.L. Circulating Expression Level of LncRNA Malat1 in Diabetic Kidney Disease Patients and Its Clinical Significance. J. Diabetes Res. 2020, 2020, 4729019. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.T.; Clark, I.M.; Garcia, P.L. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000, 58, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Jaffe, J.; Michael, M.; Swan, C.E.; Duffy, K.J.; Erickson-Miller, C.L.; Haase, V.H. Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am. J. Physiol. Renal Physiol. 2012, 302, F1172–F1179. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Matsumoto, M.; Inagi, R.; Miyata, T.; Kojima, I.; Ohse, T.; Fujita, T.; Nangaku, M. Induction of protective genes by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis. Kidney Int. 2005, 68, 2714–2725. [Google Scholar] [CrossRef]
- Viret, J.F.; Bruderer, U.; Lang, A.B. Characterization of the Shigella serotype D (S. sonnei) O polysaccharide and the enterobacterial R1 lipopolysaccharide core by use of mouse monoclonal antibodies. Infect. Immun. 1992, 60, 2741–2747. [Google Scholar] [CrossRef]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef]
- Koshiji, M.; Huang, L.E. Dynamic balancing of the dual nature of HIF-1alpha for cell survival. Cell Cycle 2004, 3, 853–854. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Fang, Y.; Ding, X.; Liu, H.; Zhu, J.; Zou, J.; Xu, X.; Zhong, Y. Transient hypoxia-inducible factor activation in rat renal ablation and reduced fibrosis with L-mimosine. Nephrology 2012, 17, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Tanaka, S.; Tanaka, T.; Saito, H.; Ishimoto, Y.; Wakashima, T.; Ueda, M.; Fukui, K.; Shimizu, A.; Inagi, R.; et al. Prolyl Hydroxylase Domain Inhibitor Protects against Metabolic Disorders and Associated Kidney Disease in Obese Type 2 Diabetic Mice. J. Am. Soc. Nephrol. 2020, 31, 560–577. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M.; Vlassara, H.; Cerami, A. Nonenzymatic glycosylation and the pathogenesis of diabetic complications. Ann. Intern. Med. 1984, 101, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Mallipattu, S.K.; Uribarri, J. Advanced glycation end product accumulation: A new enemy to target in chronic kidney disease? Curr. Opin. Nephrol. Hypertens. 2014, 23, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Saulnier, P.J.; Wheelock, K.M.; Howell, S.; Weil, E.J.; Tanamas, S.K.; Knowler, W.C.; Lemley, K.V.; Mauer, M.; Yee, B.; Nelson, R.G.; et al. Advanced Glycation End Products Predict Loss of Renal Function and Correlate with Lesions of Diabetic Kidney Disease in American Indians with Type 2 Diabetes. Diabetes 2016, 65, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kato, I.; Doi, T.; Yonekura, H.; Ohashi, S.; Takeuchi, M.; Watanabe, T.; Yamagishi, S.; Sakurai, S.; Takasawa, S.; et al. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J. Clin. Investig. 2001, 108, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.H.; Humpert, P.M.; Nawroth, P.P.; Bierhaus, A. Reactive metabolites and AGE/RAGE-mediated cellular dysfunction affect the aging process: A mini-review. Gerontology 2011, 57, 435–443. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef]
- Rabbani, N.; Alam, S.S.; Riaz, S.; Larkin, J.R.; Akhtar, M.W.; Shafi, T.; Thornalley, P.J. High-dose thiamine therapy for patients with type 2 diabetes and microalbuminuria: A randomised, double-blind placebo-controlled pilot study. Diabetologia 2009, 52, 208–212. [Google Scholar] [CrossRef]
- Bolton, W.K.; Cattran, D.C.; Williams, M.E.; Adler, S.G.; Appel, G.B.; Cartwright, K.; Foiles, P.G.; Freedman, B.I.; Raskin, P.; Ratner, R.E.; et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am. J. Nephrol. 2004, 24, 32–40. [Google Scholar] [CrossRef]
- Alkhalaf, A.; Klooster, A.; van Oeveren, W.; Achenbach, U.; Kleefstra, N.; Slingerland, R.J.; Mijnhout, G.S.; Bilo, H.J.; Gans, R.O.; Navis, G.J.; et al. A double-blind, randomized, placebo-controlled clinical trial on benfotiamine treatment in patients with diabetic nephropathy. Diabetes Care 2010, 33, 1598–1601. [Google Scholar] [CrossRef]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef]
- Oba, S.; Ayuzawa, N.; Nishimoto, M.; Kawarazaki, W.; Ueda, K.; Hirohama, D.; Kawakami-Mori, F.; Shimosawa, T.; Marumo, T.; Fujita, T. Aberrant DNA methylation of Tgfb1 in diabetic kidney mesangial cells. Sci. Rep. 2018, 8, 16338. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Kaestner, K.H.; Natarajan, R.; Patti, M.E.; Sallari, R.; Sander, M.; Susztak, K. Epigenetics and Epigenomics: Implications for Diabetes and Obesity. Diabetes 2018, 67, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Marumo, T.; Yagi, S.; Kawarazaki, W.; Nishimoto, M.; Ayuzawa, N.; Watanabe, A.; Ueda, K.; Hirahashi, J.; Hishikawa, K.; Sakurai, H.; et al. Diabetes Induces Aberrant DNA Methylation in the Proximal Tubules of the Kidney. J. Am. Soc. Nephrol. 2015, 26, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Y.; Qin, H.J.; Zhang, Z.; Xu, Y.; Yang, X.C.; Zhao, D.M.; Li, X.N.; Sun, L.K. Valproate attenuates diabetic nephropathy through inhibition of endoplasmic reticulum stress-induced apoptosis. Mol. Med. Rep. 2016, 13, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Hirakawa, Y.; Kanki, Y.; Nakaki, R.; Suzuki, Y.; Tanaka, T.; Aburatani, H.; Nangaku, M. Genome-wide analysis revealed that DZNep reduces tubulointerstitial fibrosis via down-regulation of pro-fibrotic genes. Sci. Rep. 2018, 8, 3779. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, H.; Doi, S.; Nakashima, A.; Sasaki, K.; Doi, T.; Masaki, T. Inhibition of the H3K4 methyltransferase MLL1/WDR5 complex attenuates renal senescence in ischemia reperfusion mice by reduction of p16(INK4a). Kidney Int. 2019, 96, 1162–1175. [Google Scholar] [CrossRef]
- Chen, Z.; Miao, F.; Paterson, A.D.; Lachin, J.M.; Zhang, L.; Schones, D.E.; Wu, X.; Wang, J.; Tompkins, J.D.; Genuth, S.; et al. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc. Natl. Acad. Sci. USA 2016, 113, E3002–E3011. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Xia, L.; Masson, E.A.; Gui, C.; Momen, A.; Shikatani, E.A.; Husain, M.; Quaggin, S.; John, R.; Fantus, I.G. Thioredoxin-Interacting Protein Deficiency Protects against Diabetic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 2963–2977. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Chen, Z.; Genuth, S.; Paterson, A.; Zhang, L.; Wu, X.; Li, S.M.; Cleary, P.; Riggs, A.; Harlan, D.M.; et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 2014, 63, 1748–1762. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Sumanth, P.; Lanting, L.; Yuan, H.; Wang, M.; Mar, D.; Alpers, C.E.; Bomsztyk, K.; Natarajan, R. Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int. 2014, 85, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Reddy, M.A.; Deshpande, S.; Jia, Y.; Park, J.T.; Lanting, L.L.; Jin, W.; Kato, M.; Xu, Z.G.; Das, S.; et al. Epigenetic Histone Modifications Involved in Profibrotic Gene Regulation by 12/15-Lipoxygenase and Its Oxidized Lipid Products in Diabetic Nephropathy. Antioxid. Redox Signal 2016, 24, 361–375. [Google Scholar] [CrossRef]
- Mimura, I.; Tanaka, T.; Nangaku, M. Novel therapeutic strategy with hypoxia-inducible factors via reversible epigenetic regulation mechanisms in progressive tubulointerstitial fibrosis. Semin. Nephrol. 2013, 33, 375–382. [Google Scholar] [CrossRef]
- Mimura, I.; Nangaku, M.; Kanki, Y.; Tsutsumi, S.; Inoue, T.; Kohro, T.; Yamamoto, S.; Fujita, T.; Shimamura, T.; Suehiro, J.; et al. Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol. Cell Biol. 2012, 32, 3018–3032. [Google Scholar] [CrossRef]
- Mimura, I.; Hirakawa, Y.; Kanki, Y.; Kushida, N.; Nakaki, R.; Suzuki, Y.; Tanaka, T.; Aburatani, H.; Nangaku, M. Novel lnc RNA regulated by HIF-1 inhibits apoptotic cell death in the renal tubular epithelial cells under hypoxia. Physiol. Rep. 2017, 5, e13203. [Google Scholar] [CrossRef]
- Denby, L.; Baker, A.H. Targeting non-coding RNA for the therapy of renal disease. Curr. Opin. Pharmacol. 2016, 27, 70–77. [Google Scholar] [CrossRef]
- Thielmann, M.; Corteville, D.; Szabo, G.; Swaminathan, M.; Lamy, A.; Lehner, L.J.; Brown, C.D.; Noiseux, N.; Atta, M.G.; Squiers, E.C.; et al. Teprasiran, a Small Interfering RNA, for the Prevention of Acute Kidney Injury in High-Risk Patients Undergoing Cardiac Surgery: A Randomized Clinical Study. Circulation 2021, 144, 1133–1144. [Google Scholar] [CrossRef]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Putta, S.; Lanting, L.; Sun, G.; Lawson, G.; Kato, M.; Natarajan, R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Giglio, R.V.; Patti, A.M.; Rizvi, A.A.; Stoian, A.P.; Ciaccio, M.; Papanas, N.; Janez, A.; Sonmez, A.; Banach, M.; Sahebkar, A.; et al. Advances in the Pharmacological Management of Diabetic Nephropathy: A 2022 International Update. Biomedicines 2023, 11, 291. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Zhang, C. Recent Advances in the Management of Diabetic Kidney Disease: Slowing Progression. Int. J. Mol. Sci. 2024, 25, 3086. https://doi.org/10.3390/ijms25063086
Wang N, Zhang C. Recent Advances in the Management of Diabetic Kidney Disease: Slowing Progression. International Journal of Molecular Sciences. 2024; 25(6):3086. https://doi.org/10.3390/ijms25063086
Chicago/Turabian StyleWang, Na, and Chun Zhang. 2024. "Recent Advances in the Management of Diabetic Kidney Disease: Slowing Progression" International Journal of Molecular Sciences 25, no. 6: 3086. https://doi.org/10.3390/ijms25063086
APA StyleWang, N., & Zhang, C. (2024). Recent Advances in the Management of Diabetic Kidney Disease: Slowing Progression. International Journal of Molecular Sciences, 25(6), 3086. https://doi.org/10.3390/ijms25063086