
Comprehensive Research on Druggable Proteins: From PSSM to Pre-Trained Language Models

Abstract
:1. Introduction
2. Results and Discussion
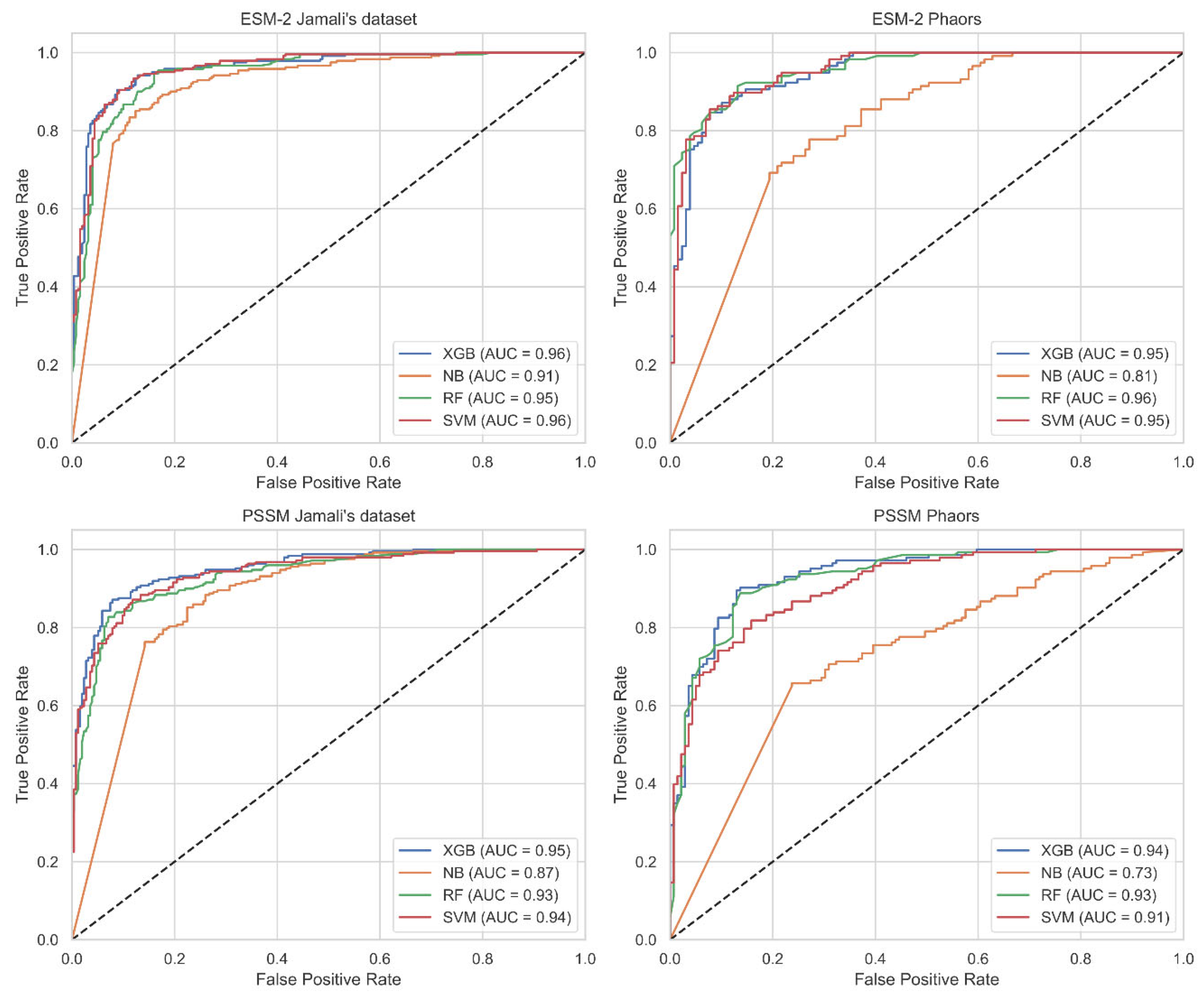
2.1. Machine Learning Classifiers Performance
2.2. Deep Learning Classifiers Performance
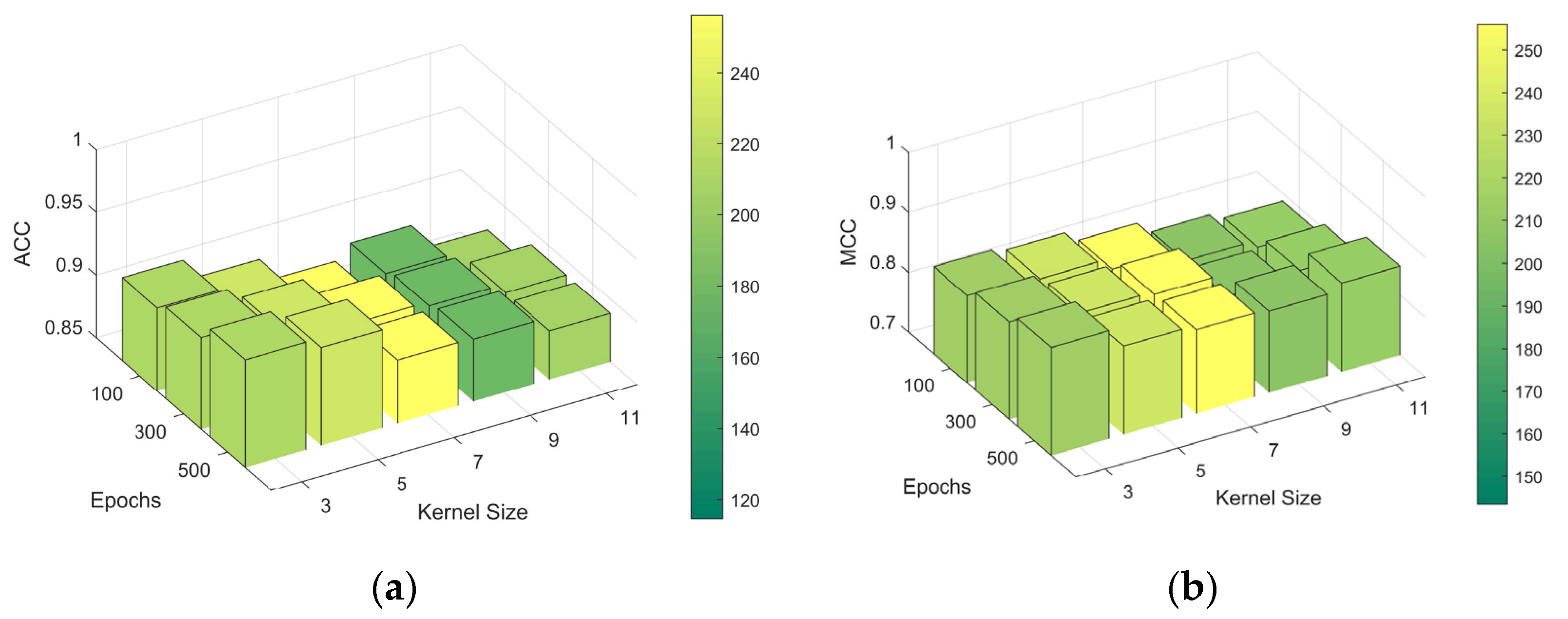
2.3. Performance Variance Analysis
2.4. Comparison with State-of-the-Art Methods
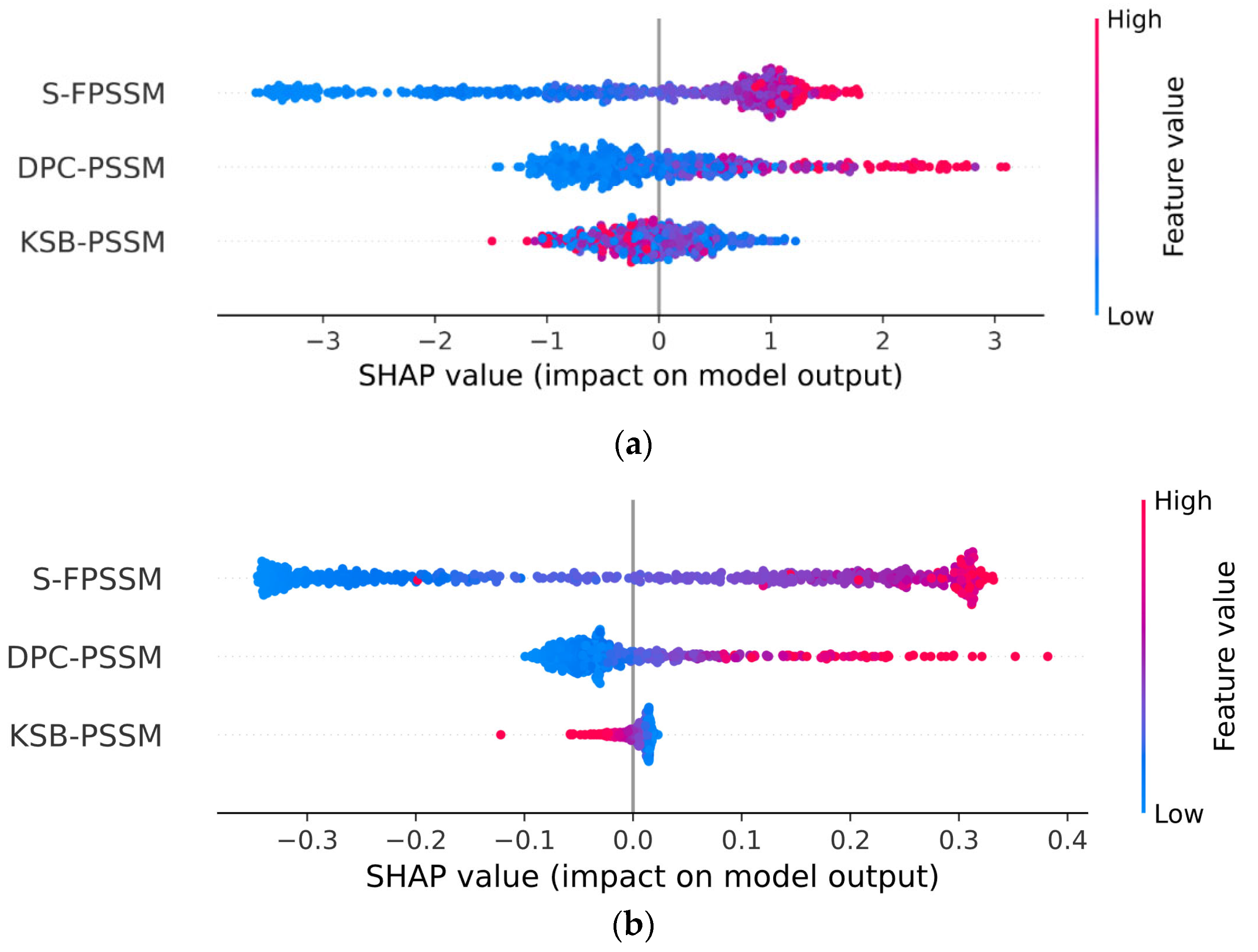
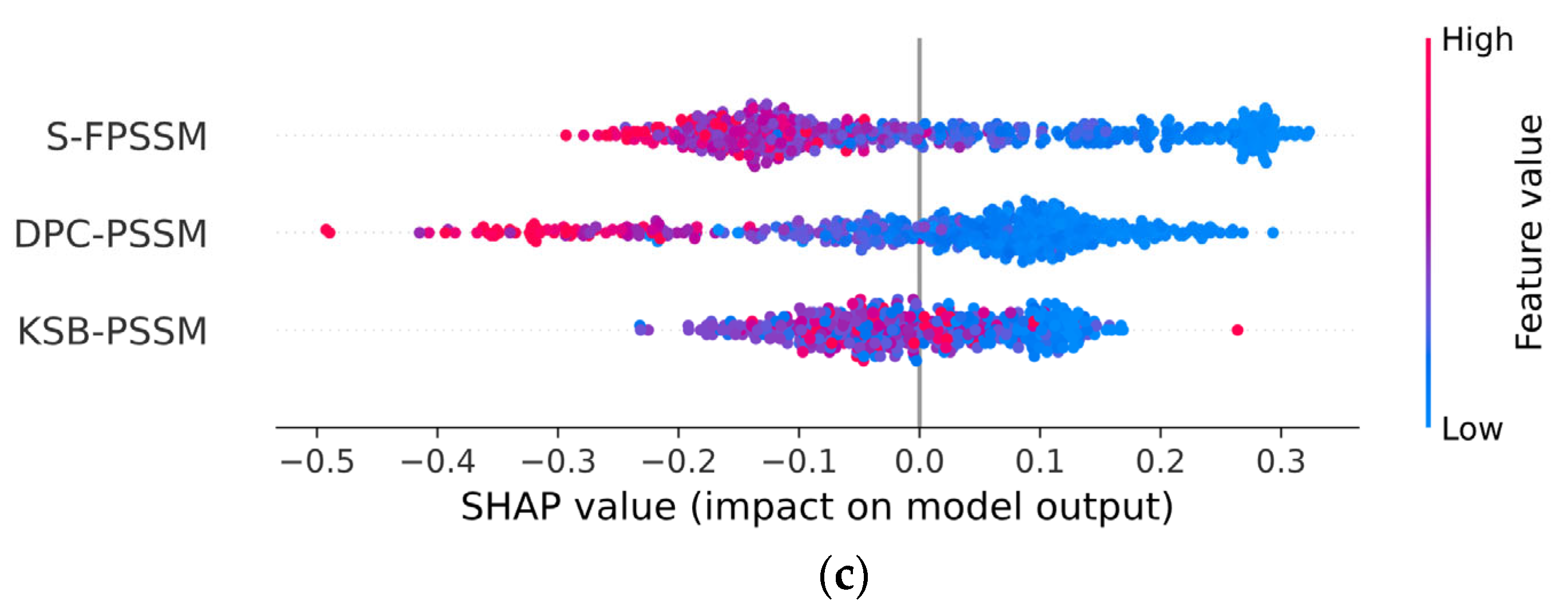
2.5. PSSM Versus PLM
2.6. Web Server
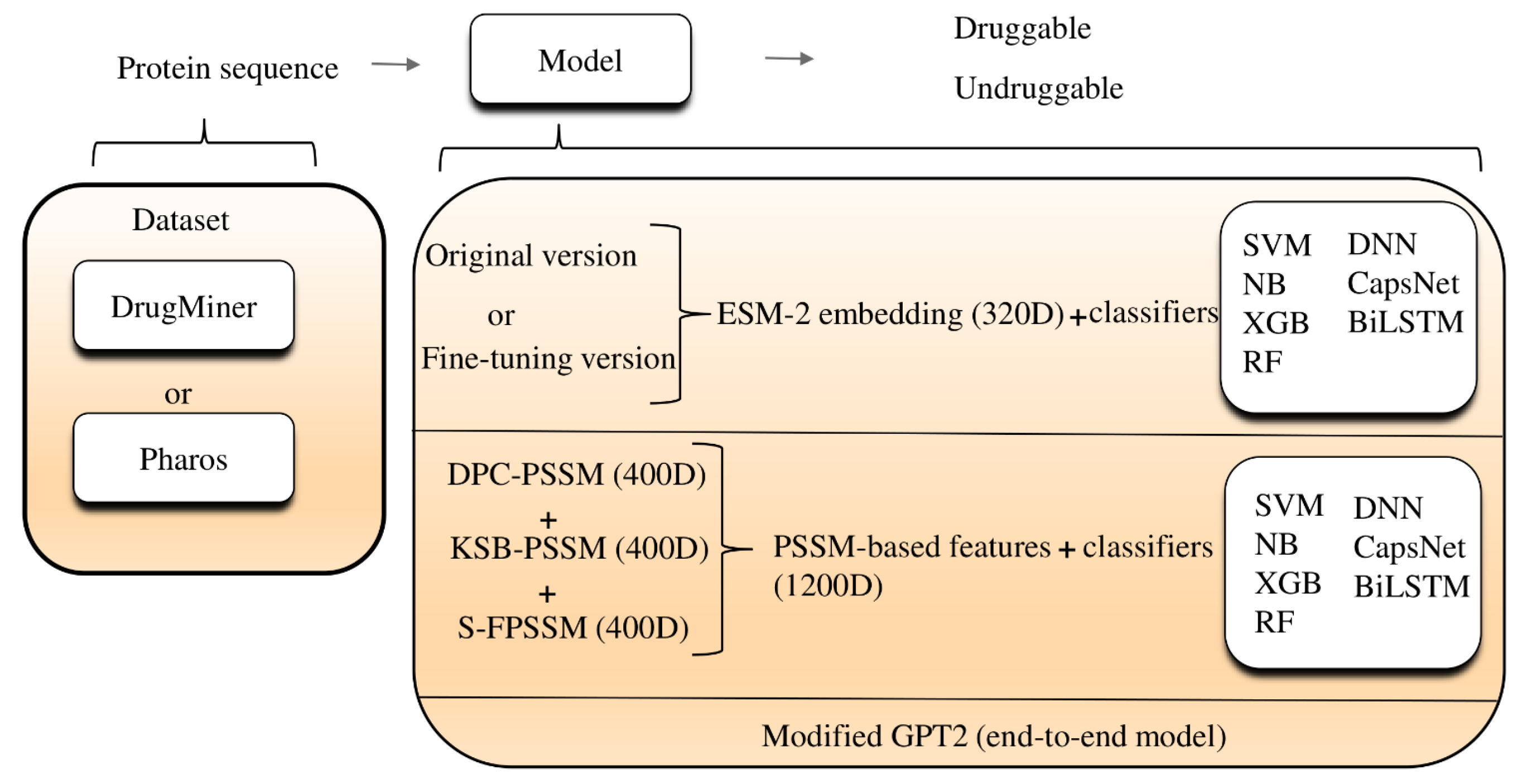
3. Materials and Methods
3.1. Datasets
3.2. Feature Representation
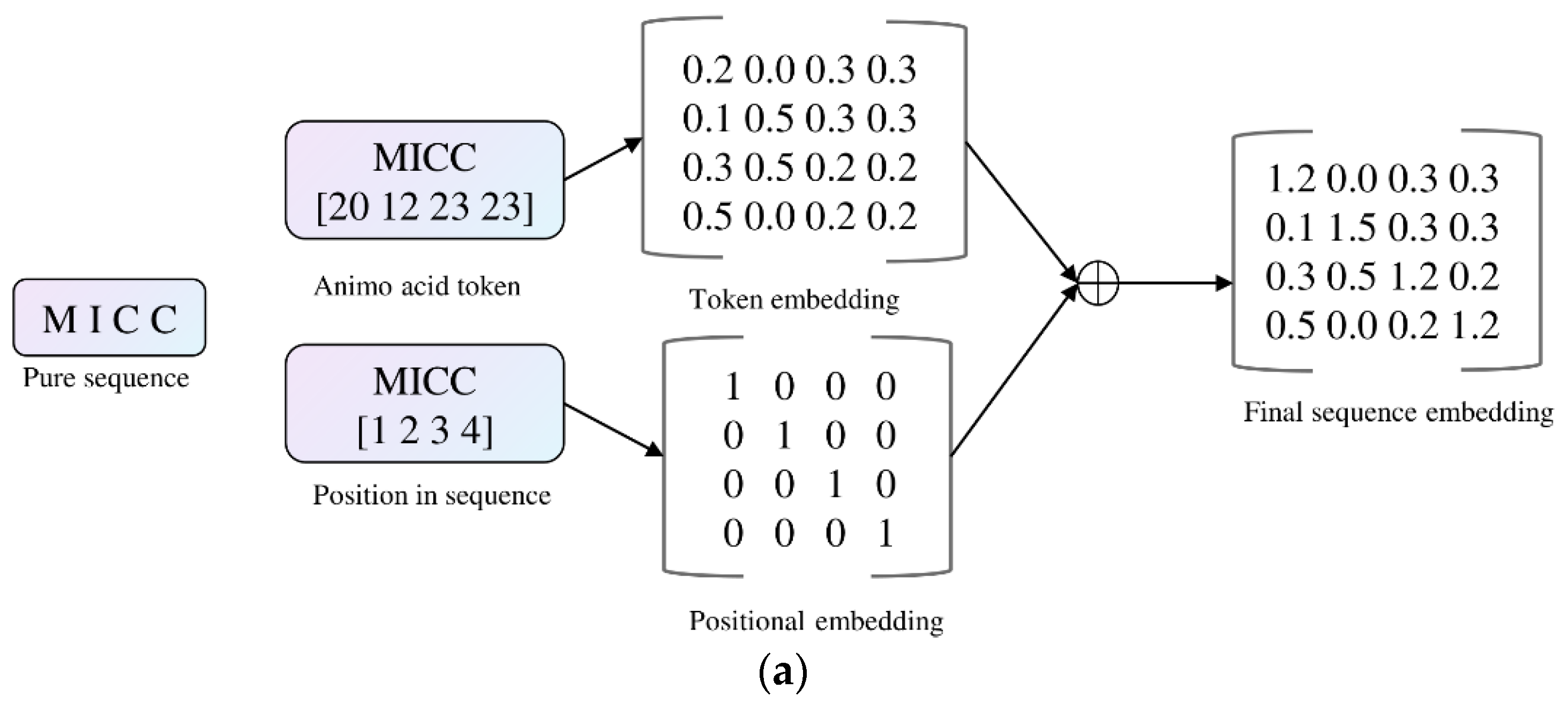
3.2.1. PLM Embeddings
3.2.2. PSSM Features
3.3. Model Architecture
3.3.1. Machine Learning Methods
3.3.2. DNN
3.3.3. CapsNet
3.3.4. BiLSTM
3.4. LLM Solution: Modified GPT-2
3.5. The Fine-Tuning Process
3.6. Performance Evaluation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Huth, J.R.; Tse, C. Predicting protein druggability. Drug Discov. Today 2005, 10, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Aguti, R.; Gardini, E.; Bertazzo, M.; Decherchi, S. Probabilistic pocket druggability prediction via one-class learning. Front. Pharmacol. 2022, 13, 870479. [Google Scholar] [CrossRef]
- Fuller, J.C.; Burgoyne, N.J.; Jackson, R.M. Predicting druggable binding sites at the protein–protein interface. Drug Discov. Today 2009, 14, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, J.; Xu, X.; Li, Y.; Zhao, H.; Fang, Y.; Li, X.; Zhou, W.; Wang, W.; Wang, Y. A systematic prediction of multiple drug-target interactions from chemical, genomic, and pharmacological data. PLoS ONE 2012, 7, e37608. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K. Advancing the drug discovery and development process. Angew. Chem. 2014, 126, 9280–9292. [Google Scholar] [CrossRef]
- Kandoi, G.; Acencio, M.L.; Lemke, N. Prediction of druggable proteins using machine learning and systems biology: A mini-review. Front. Physiol. 2015, 6, 165529. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Jamali, A.A.; Ferdousi, R.; Razzaghi, S.; Li, J.; Safdari, R.; Ebrahimie, E. DrugMiner: Comparative analysis of machine learning algorithms for prediction of potential druggable proteins. Drug Discov. Today 2016, 21, 718–724. [Google Scholar] [CrossRef]
- Lin, J.; Chen, H.; Li, S.; Liu, Y.; Li, X.; Yu, B. Accurate prediction of potential druggable proteins based on genetic algorithm and Bagging-SVM ensemble classifier. Artif. Intell. Med. 2019, 98, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Xue, L.; Liu, F.; Li, Y.; Jing, R.; Luo, J. The applications of deep learning algorithms on in silico druggable proteins identification. J. Adv. Res. 2022, 41, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hur, A.; Ong, C.S.; Sonnenburg, S.; Schölkopf, B.; Rätsch, G. Support vector machines and kernels for computational biology. PLoS Comput. Biol. 2008, 4, e1000173. [Google Scholar] [CrossRef] [PubMed]
- Alghushairy, O.; Ali, F.; Alghamdi, W.; Khalid, M.; Alsini, R.; Asiry, O. Machine learning-based model for accurate identification of druggable proteins using light extreme gradient boosting. J. Biomol. Struct. Dyn. 2023, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shi, H.; Wu, Y.; Hong, Z. Survey of natural language processing techniques in bioinformatics. Comput. Math. Methods Med. 2015, 2015, 674296. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hou, Y.; Che, W.; Liu, T. From static to dynamic word representations: A survey. Int. J. Mach. Learn. Cybern. 2020, 11, 1611–1630. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 2021, 44, 7112–7127. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef]
- Kelleher, K.J.; Sheils, T.K.; Mathias, S.L.; Yang, J.J.; Metzger, V.T.; Siramshetty, V.B.; Nguyen, D.-T.; Jensen, L.J.; Vidović, D.; Schürer, S.C. Pharos 2023: An integrated resource for the understudied human proteome. Nucleic Acids Res. 2023, 51, D1405–D1416. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Ogunleye, A.; Wang, Q.-G. XGBoost model for chronic kidney disease diagnosis. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 17, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Schuster, M.; Paliwal, K.K. Bidirectional recurrent neural networks. IEEE Trans. Signal Process. 1997, 45, 2673–2681. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Radford, A.; Wu, J.; Child, R.; Luan, D.; Amodei, D.; Sutskever, I. Language models are unsupervised multitask learners. OpenAI Blog 2019, 1, 9. [Google Scholar]
- Lundberg, S.M.; Lee, S.-I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 2017, 30. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Cunningham, M.; Pins, D.; Dezső, Z.; Torrent, M.; Vasanthakumar, A.; Pandey, A. PINNED: Identifying characteristics of druggable human proteins using an interpretable neural network. J. Cheminform. 2023, 15, 64. [Google Scholar] [CrossRef]
- Sikander, R.; Ghulam, A.; Ali, F. XGB-DrugPred: Computational prediction of druggable proteins using eXtreme gradient boosting and optimized features set. Sci. Rep. 2022, 12, 5505. [Google Scholar] [CrossRef]
- Zhang, M.; Wan, F.; Liu, T. DrugFinder: Druggable protein identification model based on pre-trained models and evolutionary information. Algorithms 2023, 16, 263. [Google Scholar] [CrossRef]
- Raies, A.; Tulodziecka, E.; Stainer, J.; Middleton, L.; Dhindsa, R.S.; Hill, P.; Engkvist, O.; Harper, A.R.; Petrovski, S.; Vitsios, D. DrugnomeAI is an ensemble machine-learning framework for predicting druggability of candidate drug targets. Commun. Biol. 2022, 5, 1291. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lai, L. Prediction of potential drug targets based on simple sequence properties. BMC Bioinform. 2007, 8, 353. [Google Scholar] [CrossRef] [PubMed]
- Bakheet, T.M.; Doig, A.J. Properties and identification of human protein drug targets. Bioinformatics 2009, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Nakamura, M.; Kajiwara, Y.; Otsuka, A.; Kimura, H. Lvq-smote–learning vector quantization based synthetic minority over–sampling technique for biomedical data. BioData Min. 2013, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Vendruscolo, M.; Kussell, E.; Domany, E. Recovery of protein structure from contact maps. Fold. Des. 1997, 2, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 1999, 27, 49–54. [Google Scholar] [CrossRef]
- Liu, T.; Zheng, X.; Wang, J. Prediction of protein structural class for low-similarity sequences using support vector machine and PSI-BLAST profile. Biochimie 2010, 92, 1330–1334. [Google Scholar] [CrossRef]
- Saini, H.; Raicar, G.; Lal, S.P.; Dehzangi, A.; Imoto, S.; Sharma, A. Protein fold recognition using genetic algorithm optimized voting scheme and profile bigram. J. Softw. 2016, 11, 756–767. [Google Scholar] [CrossRef]
- Zahiri, J.; Yaghoubi, O.; Mohammad-Noori, M.; Ebrahimpour, R.; Masoudi-Nejad, A. PPIevo: Protein–protein interaction prediction from PSSM based evolutionary information. Genomics 2013, 102, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Mao, J.; Mohiuddin, K.M. Artificial neural networks: A tutorial. Computer 1996, 29, 31–44. [Google Scholar] [CrossRef]
- Sabour, S.; Frosst, N.; Hinton, G.E. Dynamic routing between capsules. Adv. Neural Inf. Process. Syst. 2017, 30. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Graves, A. Long Short-Term Memory. Supervised Sequence Labelling with Recurrent Neural Networks; Springer: Berlin/Heidelberg, Germany, 2012; pp. 37–45. [Google Scholar]
- Rao, R.; Meier, J.; Sercu, T.; Ovchinnikov, S.; Rives, A. Transformer protein language models are unsupervised structure learners. Biorxiv 2020. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Encoding | Classifier | ACC | P | SN | SP | F1 | MCC |
|---|---|---|---|---|---|---|---|
| PSSM (1200 dimensions) | SVM | 0.8271 ±0.0169 | 0.8280 ±0.0269 | 0.8214 ±0.0327 | 0.8326 ±0.0176 | 0.8247 ±0.0263 | 0.6541 ±0.0309 |
| XGB | 0.9037 ±0.0245 | 0.9043 ±0.0203 | 0.9007 ±0.0332 | 0.9066 ±0.0146 | 0.9025 ±0.0200 | 0.8074 ±0.0212 | |
| NB | 0.7347 ±0.0295 | 0.8232 ±0.0503 | 0.5912 ±0.0646 | 0.8754 ±0.0256 | 0.6882 ±0.0471 | 0.4875 ±0.0597 | |
| RF | 0.8664 ±0.0269 | 0.8593 ±0.0341 | 0.8730 ±0.0220 | 0.8599 ±0.0169 | 0.8661 ±0.0268 | 0.7329 ±0.0276 | |
| ESM-2 (320 dimensions) | SVM | 0.9326 ±0.0087 | 0.9406 ±0.0121 | 0.9196 ±0.0114 | 0.9449 ±0.0098 | 0.9300 ±0.0158 | 0.8652 ±0.0154 |
| XGB | 0.9065 ±0.0170 | 0.8951 ±0.0179 | 0.9151 ±0.0246 | 0.8983 ±0.0114 | 0.9050 ±0.0289 | 0.8132 ±0.0369 | |
| NB | 0.8847 ±0.0153 | 0.8904 ±0.0156 | 0.8705 ±0.0247 | 0.8983 ±0.0250 | 0.8803 ±0.0258 | 0.7694 ±0.0289 | |
| RF | 0.8913 ±0.0177 | 0.8990 ±0.0237 | 0.8750 ±0.0331 | 0.9067 ±0.0127 | 0.8868 ±0.0135 | 0.7825 ±0.0318 |
| Encoding | Classifier | ACC | P | SN | SP | F1 | MCC |
|---|---|---|---|---|---|---|---|
| PSSM (1200 dimensions) | SVM | 0.8156 ±0.0296 | 0.8527 ±0.0287 | 0.7692 ±0.0326 | 0.8633 ±0.0135 | 0.8088 ±0.0311 | 0.6347 ±0.0470 |
| XGB | 0.8226 ±0.0254 | 0.8842 ±0.0198 | 0.7482 ±0.0343 | 0.8992 ±0.0121 | 0.8106 ±0.0212 | 0.6540 ±0.0429 | |
| NB | 0.7234 ±0.0356 | 0.7731 ±0.0301 | 0.6433 ±0.0431 | 0.8057 ±0.0198 | 0.7022 ±0.0232 | 0.4546 ±0.0553 | |
| RF | 0.8120 ±0.0155 | 0.8947 ±0.0217 | 0.7132 ±0.0236 | 0.9136 ±0.0292 | 0.7937 ±0.0252 | 0.6387 ±0.0370 | |
| ESM-2 (320 dimensions) | SVM | 0.8455 ±0.0285 | 0.8264 ±0.0352 | 0.8547 ±0.0126 | 0.8372 ±0.0194 | 0.8403 ±0.0356 | 0.6911 ±0.0419 |
| XGB | 0.8739 ±0.0192 | 0.8208 ±0.0148 | 0.9401 ±0.0150 | 0.8139 ±0.0236 | 0.8764 ±0.0142 | 0.7562 ±0.0359 | |
| NB | 0.7195 ±0.0257 | 0.6578 ±0.0411 | 0.8547 ±0.0266 | 0.5968 ±0.0303 | 0.7434 ±0.0325 | 0.4641 ±0.0606 | |
| RF | 0.8739 ±0.0211 | 0.8307 ±0.0157 | 0.9207 ±0.0305 | 0.8294 ±0.0359 | 0.8749 ±0.0167 | 0.7528 ±0.0227 |
| Encoding | Classifier | ACC | P | SN | SP | F1 | MCC |
|---|---|---|---|---|---|---|---|
| PSSM (1200 dimensions) | DNN | 0.8827 ±0.0134 | 0.9318 ±0.0162 | 0.8233 ±0.0139 | 0.9409 ±0.0198 | 0.8742 ±0.0271 | 0.7703 ±0.0347 |
| CapsNet | 0.8740 ±0.0173 | 0.8245 ±0.0284 | 0.9495 ±0.0147 | 0.7989 ±0.0277 | 0.8826 ±0.0137 | 0.7568 ±0.0276 | |
| BiLSTM | 0.8867 ±0.0144 | 0.9138 ±0.0167 | 0.8514 ±0.0229 | 0.9213 ±0.0226 | 0.8815 ±0.0125 | 0.7750 ±0.0304 | |
| ESM-2 (320 dimensions) | DNN | 0.9105 ±0.0160 | 0.8991 ±0.0141 | 0.9145 ±0.0153 | 0.9069 ±0.0297 | 0.9067 ±0.0220 | 0.8209 ±0.0285 |
| CapsNet | 0.9340 ±0.0129 | 0.9128 ±0.0312 | 0.9544 ±0.0164 | 0.9151 ±0.0119 | 0.9331 ±0.0211 | 0.8690 ±0.0252 | |
| BiLSTM | 0.8984 ±0.0171 | 0.8736 ±0.0184 | 0.9209 ±0.0131 | 0.8778 ±0.0172 | 0.8966 ±0.0219 | 0.7979 ±0.0278 |
| Encoding | Classifier | ACC | P | SN | SP | F1 | MCC |
|---|---|---|---|---|---|---|---|
| PSSM (1200 dimensions) | DNN | 0.8886 ±0.0120 | 0.8789 ±0.0218 | 0.9074 ±0.0197 | 0.8689 ±0.0319 | 0.8929 ±0.0317 | 0.7774 ±0.0293 |
| CapsNet | 0.8602 ±0.0193 | 0.9026 ±0.0146 | 0.8148 ±0.0246 | 0.9078 ±0.0177 | 0.8564 ±0.0119 | 0.7245 ±0.0362 | |
| BiLSTM | 0.8578 ±0.0137 | 0.8333 ±0.0179 | 0.9028 ±0.0253 | 0.8107 ±0.0214 | 0.8667 ±0.0198 | 0.7175 ±0.0272 | |
| ESM-2 (320 dimensions) | DNN | 0.9037 ±0.0164 | 0.8889 ±0.0243 | 0.9119 ±0.0114 | 0.8962 ±0.0166 | 0.9003 ±0.0226 | 0.8075 ±0.0203 |
| CapsNet | 0.8997 ±0.0156 | 0.8677 ±0.0254 | 0.9318 ±0.0171 | 0.8705 ±0.0178 | 0.8986 ±0.0170 | 0.8017 ±0.0206 | |
| BiLSTM | 0.8222 ±0.0317 | 0.8870 ±0.0265 | 0.7185 ±0.0382 | 0.9167 ±0.0445 | 0.7939 ±0.0417 | 0.6516 ±0.0501 |
| Model | ACC | SN | SP | F1 | MCC |
|---|---|---|---|---|---|
| DrugMiner [10] | 0.9210 | 0.9280 | 0.9134 | 0.9241 | 0.8417 |
| GA-Bagging-SVM [11] | 0.9378 | 0.9286 | 0.9445 | 0.9358 | 0.8781 |
| XGB-DrugPred [30] | 0.9486 | 0.9375 | 0.9574 | 0.9417 | 0.8900 |
| DrugFinder [31] | 0.9498 | 0.9633 | 0.9683 | 0.9460 | 0.8996 |
| Modified GPT-2 | 0.9282 | 0.9332 | 0.9224 | 0.9332 | 0.8556 |
| Fine-tunned ESM-2 with CapsNet | 0.9511 | 0.9683 | 0.9691 | 0.9512 | 0.9011 |
| Web Server Link | Summary | Data Available |
|---|---|---|
| https://www.drugminer.org | Search for druggable protein and view their features | yes |
| http://drugnomeai.public.cgr.astrazeneca.com | Provide visualization and clear explanation of the findings | yes |
| https://druggableprotein.com (ours) | Upload files in FASTA format and receive predicted results | yes |
| Dataset | Train Positive | Train Negative | Validation Positive | Validation Negative | Test Positive | Test Negative |
|---|---|---|---|---|---|---|
| Jamali’s | 784 | 845 | 196 | 211 | 244 | 263 |
| Pharos | 452 | 452 | 112 | 112 | 140 | 140 |
| Dataset | Longest | Shortest | Mean | Medium | Standard Deviation |
|---|---|---|---|---|---|
| Jamali’s | 5762 | 8 | 506 | 390 | 521 |
| Pharos | 34,350 | 2 | 554 | 411 | 528 |
| Model | Final Hyperparameters |
|---|---|
| SVM | C = 10, gamma = ’scale’, decision_function_shape = ’ovr’, kernel = ’rbf’ |
| RF | n_estimators = 1000, max_depth = 3, random_state = 0, n_jobs = −1 |
| NB | priors = None, var_smoothing = 1 × 10−9 |
| XGB | max_depth = 15, learning_rate = 0.1, n_estimators = 2000, min_child_weight = 5, max_delta_step = 0, subsample = 0.8, colsample_bytree = 0.7, reg_alpha = 0, reg_lambda = 0.4, scale_pos_weight = 0.8, objective = ’binary:logistic’, eval_metric = ’auc’, seed = 1440, gamma = 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, H.; Liu, T. Comprehensive Research on Druggable Proteins: From PSSM to Pre-Trained Language Models. Int. J. Mol. Sci. 2024, 25, 4507. https://doi.org/10.3390/ijms25084507
Chu H, Liu T. Comprehensive Research on Druggable Proteins: From PSSM to Pre-Trained Language Models. International Journal of Molecular Sciences. 2024; 25(8):4507. https://doi.org/10.3390/ijms25084507
Chicago/Turabian StyleChu, Hongkang, and Taigang Liu. 2024. "Comprehensive Research on Druggable Proteins: From PSSM to Pre-Trained Language Models" International Journal of Molecular Sciences 25, no. 8: 4507. https://doi.org/10.3390/ijms25084507
APA StyleChu, H., & Liu, T. (2024). Comprehensive Research on Druggable Proteins: From PSSM to Pre-Trained Language Models. International Journal of Molecular Sciences, 25(8), 4507. https://doi.org/10.3390/ijms25084507