

Integrative and Conjugative Elements and Prophage DNA as Carriers of Resistance Genes in Erysipelothrix rhusiopathiae Strains from Domestic Geese in Poland

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Identification and Serotyping
2.2. Basic Genomic Analyses
2.3. Antibiotic Susceptibility and Resistance Gene Profiles
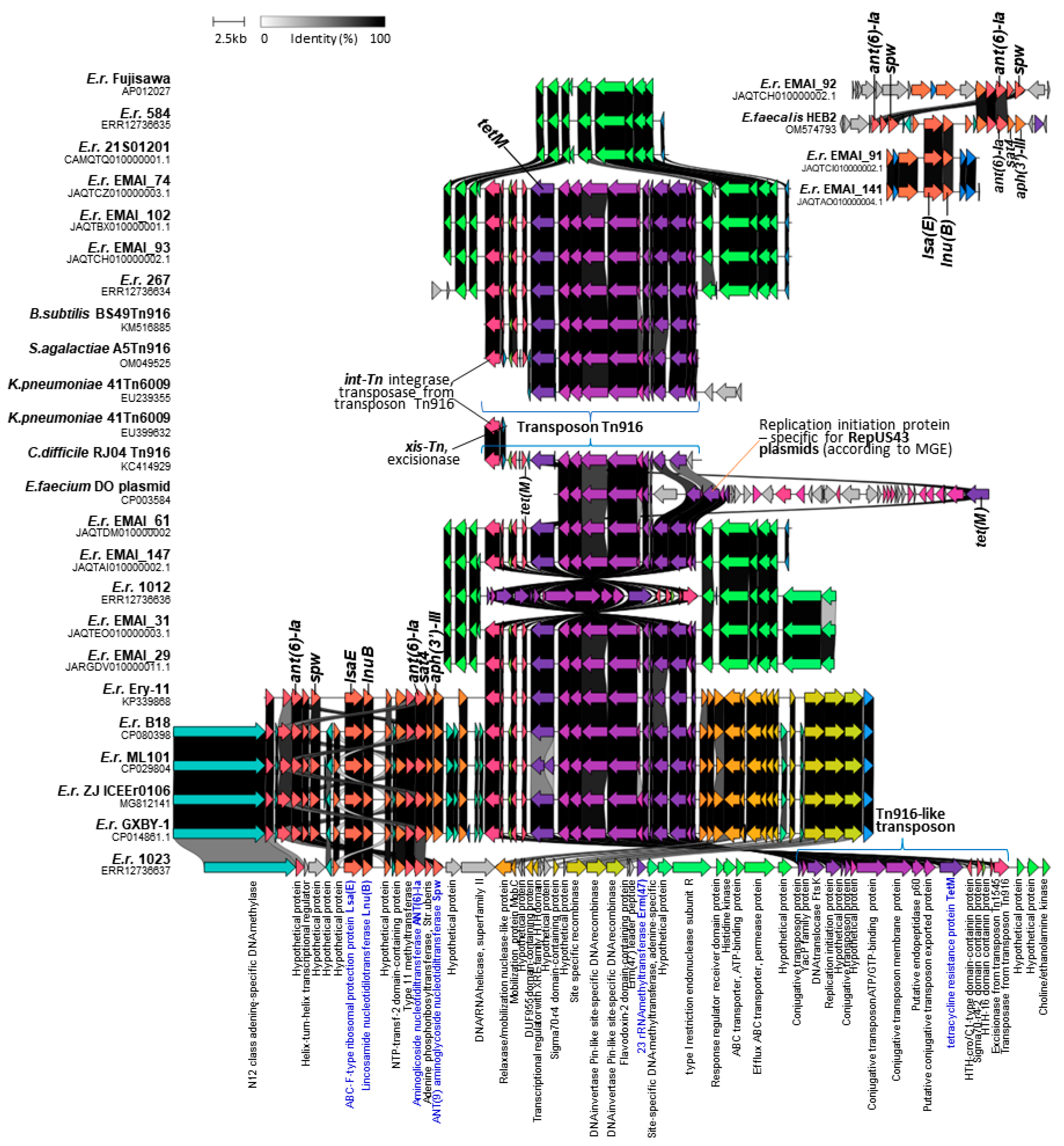
2.4. Identification of ICEs
2.5. Detection of Phage DNA and Its Possible Involvement in the Transduction of Resistance
2.6. MLST Results
2.7. Phylogenetic Inference
3. Materials and Methods
3.1. Isolation, Identification, and Phenotypic Characterization of E. rhusiopathiae Strains
3.2. Whole-Genome Sequencing
3.3. WGSs Used in Comparative Analysis and Determination of Homology between DNA Sequences
3.4. Multilocus Sequence Typing
3.5. Detection of Resistance Genes, Mobile Genetic Elements, and Phage DNA
3.6. Phylogenetic Inference
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain ID | Prophage DNA Location | Length [kb] | Score * | Total Proteins | Most Common Phage | GC Content | Presence of Resistance Genes |
|---|---|---|---|---|---|---|---|
| 267 | 352550…374967 | 22.4 | 50 | 26 | PHAGE_Erysip_SE_1 NC_029078(6) | 37.51% | No |
| 905922…913616 | 7.6 | 30 | 10 | PHAGE_Geobac_GBSV1 NC_008376(2) | 42.38% | ermB was found next to phage DNA (914 218…914 955; length 738 bp) | |
| 584 | 617896…660749 | 42.8 | 130 | 43 | PHAGE_Erysip_SE_1 NC_029078(34) | 35.13% | NA |
| 548403…566281 | 17.8 | 50 | 22 | PHAGE_Erysip_SE_1 NC_029078(6) | 37.63% | NA | |
| 1012 | 1614477…1637026 | 22.5 | 60 | 26 | PHAGE_Erysip_SE_1 NC_029078(6) | 37.45% | No |
| 1023 | 709331…735594 | 26.2 | 20 | 21 | PHAGE_Bacill_BCJA1c NC_006557(6) | 34.68 | No |
| CTCTTATTGATAAAGTCGTATTGAACACCTAAGTCGATTACTTCACCAATATAGCTAATTCCCTTACCGTAAATAATTTCAACTTGTGTTGCT CTAAAGGGCGGTGCCACTTTATTCTTTACGACTTTAATATTAGCTTTATTACCCACAATATCTGTACCTTGTTTTATTTGTTCACTACGACGA ATATCAAGTCGAACAGAAGAATAGAATTTTAAGGCACGTCCTCCCGGTGTTGTTTCAGGATTACCAAACATAATACCTACTTTTTCTCGA AGTTGATTAATAAAAATTGCAGTACACTCGCCTCGGTTCATTCCACCGGATAATTTACGCATTGCTTTAGACATCATCCGAGCTTGTAAAC CAACTTGAGCATCGCCCATTTCCCCATCAAGTTCCGCTTGTGGAACAAGTGCAGCAACACTGTCCACCACAATCAAGTCAACCGCACCAC TTCGTACCAAAACATCCACAATTTCCAATCCTTGTTCACCGCTATCGGGTTGTGAAAGAATTAAATCATCAATGTTAACGCCTAAGTTTTGA GCATAGATAGGATCAATCGCGTTCTCAGCATCGATAAATGCAGCTTTTCCACCCGCTTTTTGCACTTCTGCAATTGCATGTAATGCGAGCG |
References
- Zautner, A.E.; Tersteegen, A.; Schiffner, C.J.; Ðilas, M.; Marquardt, P.; Riediger, M.; Delker, A.M.; Mäde, D.; Kaasch, A.J. Human Erysipelothrix rhusiopathiae infection via bath water—Case report and genome announcement. Front. Cell Infect. Microbiol. 2022, 12, 981477. [Google Scholar] [CrossRef] [PubMed]
- Dec, M.; Łagowski, D.; Nowak, T.; Pietras-Ożga, D.; Herman, K. Serotypes, Antibiotic Susceptibility, Genotypic Virulence Profiles and SpaA Variants of Erysipelothrix rhusiopathiae Strains Isolated from Pigs in Poland. Pathogens 2023, 12, 409. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E. Erysipelothrix. In Bergey’s Manual of Systematics of Archaea and Bacteria, 1st ed.; Whitman, W.B., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA; Bergey’s Manual Trust: Glasgow, UK, 2015; pp. 1–15. [Google Scholar] [CrossRef]
- IJsseldijk, L.L.; Begeman, L.; Duim, B.; Gröne, A.; Kik, M.J.L.; Klijnstra, M.D.; Lakemeyer, J.; Leopold, M.; Munnink, B.B.O.; Ten Doeschate, M.; et al. Harbor Porpoise Deaths Associated with Erysipelothrix rhusiopathiae, the Netherlands, 2021. Emerg. Infect. Dis. 2023, 29, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Bender, J.S.; Shen, H.G.; Irwin, C.K.; Schwartz, K.J.; Opriessnig, T. Characterization of Erysipelothrix species isolates from clinically affected pigs, environmental samples, and vaccine strains from six recent swine erysipelas outbreaks in the United States. Clin. Vaccine Immunol. 2010, 17, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.L.; Harrington, R., Jr. Serotypes of Erysipelothrix rhusiopathiae isolated from swine and from soil and manure of swine pens in the United States. Am. J. Vet. Res. 1978, 39, 1833–1840. [Google Scholar] [PubMed]
- Ding, Y.; Zhu, D.; Zhang, J.; Yang, L.; Wang, X.; Chen, H.; Tan, C. Virulence determinants, antimicrobial susceptibility, and molecular profiles of Erysipelothrix rhusiopathiae strains isolated from China. Emerg. Microbes Infect. 2015, 4, e69. [Google Scholar] [CrossRef] [PubMed]
- Dessalew, H.; Dessalew, T.; Tadesse, T. Swine Erysipelas: It’s Epidemiology, Diagnosis, Treatment, Control, Preventive Measures and Comprehensive Review. J. Vet. Sci. Technol. 2021, 12, 100. [Google Scholar]
- Meier, S.M.; Kottwitz, J.; Keller, D.I.; Albini, S. Erysipelothrix rhusiopathiae infection by geese to human transmission. BMJ Case Rep. 2021, 14, e240073. [Google Scholar] [CrossRef] [PubMed]
- Bobrek, K.; Nowak, M.; Borkowska, J.; Bobusia, K.; Gaweł, A. An outbreak of erysipelas in commercial geese. Pak. Vet. J. 2016, 36, 372–374. [Google Scholar]
- Jordan, F.T.W.; Bisgaard, M. Erysipelothrix rhusiopathiae—różyca. In Choroby Drobiu, ; Pattison, M., McMullin, P.F., Bradbury, J.M., Alexander, D.J., Wieliczko, A., Eds.; Edra Urban & Partner: Wrocław, Poland, 2011; pp. 257–261. (In Polish)rhusiopathiae—różyca. In Choroby Drobiu, 2011, 1st ed.; Pattison, M., McMullin, P.F., Bradbury, J.M., Alexander, D.J., Wieliczko, A., Eds.; Edra Urban & Partner: Wrocław, Poland, 2011; pp. 257–261. (In Polish) [Google Scholar]
- Bobrek, K.; Gaweł, A. Antimicrobial Resistance of Erysipelothrix rhusiopathiae Strains Isolated from Geese to Antimicrobials Widely Used in Veterinary Medicine. Antibiotics 2023, 12, 1339. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lv, C.; Zhao, Y.; Zhu, W.; Liu, L.; Wang, T.; Kang, C.; Yang, Y.; Sun, X.; Zhang, Q.; et al. Characterization of Erysipelothrix Rhusiopathiae Isolates from Diseased Pigs in 15 Chinese Provinces from 2012 to 2018. Microorganisms 2021, 9, 2615. [Google Scholar] [CrossRef]
- Gu, J.; Li, Y.X.; Xu, C.W.; Xie, X.J.; Li, P.; Ma, G.X.; Lei, C.W.; Liu, J.X.; Zhang, A.Y. Genome sequence of multidrug-resistant Erysipelothrix rhusiopathiae ZJ carrying several acquired antimicrobial resistance genes. J. Glob. Antimicrob. Resist. 2020, 21, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhu, Y.; Peng, Z.; Ding, Y.; Jie, K.; Wang, Z.; Peng, Y.; Tang, X.; Wang, X.; Chen, H.; et al. Comparative Genome Analysis of a Pathogenic Erysipelothrix Rhusiopathiae Isolate WH13013 from Pig Reveals Potential Genes Involve in Bacterial Adaptions and Pathogenesis. Vet. Sci. 2020, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Ooka, T.; Shi, F.; Ogura, Y.; Nakayama, K.; Hayashi, T.; Shimoji, Y. The genome of Erysipelothrix rhusiopathiae, the causative agent of swine erysipelas, reveals new insights into the evolution of firmicutes and the organism’s intracellular adaptations. J. Bacteriol. 2011, 193, 2959–2971. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.B.; Xie, J.; Wang, L.; Liu, F.; Wu, J. Complete genome sequence of Erysipelothrix rhusiopathiae strain GXBY-1 isolated from acute swine erysipelas outbreaks in south China. Genom. Data 2016, 8, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, N.; Sasatsu, M.; Takahashi, T.; Ohmae, K.; Terakado, N.; Kono, M. Detection of plasmid DNA in Erysipelothrix rhusiopathiae isolated from pigs with chronic swine erysipelas. J. Vet. Med. Sci. 1993, 55, 349–350. [Google Scholar] [CrossRef] [PubMed]
- Pomerantsev, A.P.; Manzeniuk, I.N.; Svetoch, T.E.; Stepanshin, I.G.; Kondruk, E.K.; Gusev, V.V.; Svetoch, E.A. Erysipelothrix rhusiopathiae: Plasmidy, ustoĭchivost’ k antibakterial’nym preparatam [Erysipelothrix rhusiopathiae: Plasmids, resistance to antibacterial drugs]. Antibiot. Khimioter. 1996, 41, 30–35. (In Russian) [Google Scholar] [PubMed]
- Xu, C.W.; Zhang, A.Y.; Yang, C.M.; Pan, Y.; Guan, Z.B.; Lei, C.W.; Peng, L.Y.; Li, Q.Z.; Wang, H.N. First report of macrolide resistance gene erm(T) harbored by a novel small plasmid from Erysipelothrix rhusiopathiae. Antimicrob. Agents Chemother. 2015, 59, 2462–2465. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, T.A.; Imada, Y.; Barcellos, D.E.S.N.; Oliveira, S.J.; Moreno, A.M. Phenotypic and Molecular Characterization of Recent and Archived Erysipelothrix Spp. Isolated from Brazilian Swine. Diagn. Microbiol. Infect. Dis. 2011, 69, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Petinaki, E.; Guérin-Faublée, V.; Pichereau, V.; Villers, C.; Achard, A.; Malbruny, B.; Leclercq, R. Lincomycin resistance gene lnu(D) in Streptococcus uberis. Antimicrob. Agents Chemother. 2008, 52, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Chuma, T.; Kawamoto, T.; Shahada, F.; Fujimoto, H.; Okamoto, K. Antimicrobial susceptibility of Erysipelothrix rhusiopathiae isolated from pigs in Southern Japan with a modified agar dilution method. J. Vet. Med. Sci. 2010, 72, 643–645. [Google Scholar] [CrossRef]
- Hess, C.; Bilic, I.; Jandreski-Cvetkovic, D.; Hess, M. Antimicrobial Dilution Susceptibility Testing of Erysipelothrix rhusiopathiae According to CLSI Document VET06 Reveals High Resistance against Penicillin G, Erythromycin and Enrofloxacin. Poultry 2023, 2, 54–62. [Google Scholar] [CrossRef]
- Guérin, F.; Isnard, C.; Bucquet, F.; Fines-Guyon, M.; Giard, J.C.; Burrus, V.; Cattoir, V. Novel chromosome-encoded erm(47) determinant responsible for constitutive MLSB resistance in Helcococcus kunzii. J. Antimicrob. Chemother. 2016, 71, 3046–3049. [Google Scholar] [CrossRef]
- Tomazetto, G.; Hahnke, S.; Maus, I.; Wibberg, D.; Pühler, A.; Schlüter, A.; Klocke, M. Complete genome sequence of Peptoniphilus sp. strain ING2-D1G isolated from a mesophilic lab-scale completely stirred tank reactor utilizing maize silage in co-digestion with pig and cattle manure for biomethanation. J. Biotechnol. 2014, 192 Pt A, 59–61. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, K.; Du, X.; Lu, Y.; Liao, L.; He, Z.; He, W. Translational Attenuation Mechanism of ErmB Induction by Erythromycin Is Dependent on Two Leader Peptides. Front. Microbiol. 2021, 12, 690744. [Google Scholar] [CrossRef]
- Chen, L.; Huang, J.; Huang, X.; He, Y.; Sun, J.; Dai, X.; Wang, X.; Shafiq, M.; Wang, L. Horizontal Transfer of Different erm(B)-Carrying Mobile Elements Among Streptococcus suis Strains with Different Serotypes. Front. Microbiol. 2021, 12, 628740. [Google Scholar] [CrossRef]
- Urban-Chmiel, R.; Marek, A.; Stępień-Pyśniak, D.; Wieczorek, K.; Dec, M.; Nowaczek, A.; Osek, J. Antibiotic Resistance in Bacteria—A Review. Antibiotics 2022, 11, 1079. [Google Scholar] [CrossRef]
- Dec, M.; Stępień-Pyśniak, D.; Nowaczek, A.; Puchalski, A.; Urban-Chmiel, R. Phenotypic and genotypic antimicrobial resistance profiles of fecal lactobacilli from domesticated pigeons in Poland. Anaerobe 2020, 65, 102251. [Google Scholar] [CrossRef]
- Flannagan, S.E.; Zitzow, L.A.; Su, Y.A.; Clewell, D.B. Nucleotide sequence of the 18-kb conjugative transposon Tn916 from Enterococcus faecalis. Plasmid 1994, 32, 350–354. [Google Scholar] [CrossRef]
- Browne, H.P.; Anvar, S.Y.; Frank, J.; Lawley, T.D.; Roberts, A.P.; Smits, W.K. Complete genome sequence of BS49 and draft genome sequence of BS34A, Bacillus subtilis strains carrying Tn916. FEMS Microbiol. Lett. 2015, 362, 1–4. [Google Scholar] [CrossRef]
- Dong, D.; Chen, X.; Jiang, C.; Zhang, L.; Cai, G.; Han, L.; Wang, X.; Mao, E.; Peng, Y. Genetic analysis of Tn916-like elements conferring tetracycline resistance in clinical isolates of Clostridium difficile. Int. J. Antimicrob. Agents. 2014, 43, 73–77. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Ruangchai, W.; Thawornwattana, Y.; Jaemsai, B.; Mahasirimongkol, S.; Homkaew, A.; Suksomchit, P.; Dubbs, P.; Palittapongarnpim, P. Genomic epidemiology of Streptococcus agalactiae ST283 in Southeast Asia. Sci. Rep. 2022, 12, 4185. [Google Scholar] [CrossRef]
- Soge, O.O.; Beck, N.K.; White, T.M.; No, D.B.; Roberts, M.C. A novel transposon, Tn6009, composed of a Tn916 element linked with a Staphylococcus aureus mer operon. J. Antimicrob. Chemother. 2008, 62, 674–680. [Google Scholar] [CrossRef]
- Roberts, A.P.; Mullany, P. Tn916-like genetic elements: A diverse group of modular mobile elements conferring antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 856–871. [Google Scholar] [CrossRef]
- Taylor, K.L.; Churchward, G. Specific DNA cleavage mediated by the integrase of conjugative transposon Tn916. J. Bacteriol. 1997, 179, 1117–1125. [Google Scholar] [CrossRef]
- Mullany, P.R.; Williams, G.C.; Langridge, D.J.; Turner, R.; Whalan, C.; Clayton, T.; Lawley, H.; Hussain, K.; Mccurrie, N.; Morden, E.; et al. Behavior and target site selection of conjugative transposon Tn916 in two different strains of toxigenic Clostridium difficile. Appl. Environ. Microbiol. 2012, 78, 2147–2153. [Google Scholar] [CrossRef]
- Qin, X.; Galloway-Peña, J.R.; Sillanpaa, J.; Roh, J.H.; Nallapareddy, S.R.; Chowdhury, S.; Bourgogne, A.; Choudhury, T.; Muzny, D.M.; Buhay, C.J.; et al. Complete genome sequence of Enterococcus faecium strain TX16 and comparative genomic analysis of Enterococcus faecium genomes. BMC Microbiol. 2012, 12, 135. [Google Scholar] [CrossRef]
- Lin, I.H.; Liu, T.T.; Teng, Y.T.; Wu, H.L.; Liu, Y.M.; Wu, K.M.; Chang, C.H.; Hsu, M.T. Sequencing and comparative genome analysis of two pathogenic Streptococcus gallolyticus subspecies: Genome plasticity, adaptation and virulence. PLoS ONE 2011, 6, e20519. [Google Scholar] [CrossRef]
- Raftis, E.J.; Forde, B.M.; Claesson, M.J.; O’Toole, P.W. Unusual genome complexity in Lactobacillus salivarius JCM1046. BMC Genom. 2014, 15, 771. [Google Scholar] [CrossRef]
- Johnson, C.M.; Grossman, A.D. Integrative and Conjugative Elements (ICEs): What They Do and How They Work. Annu. Rev. Genet. 2015, 49, 577–601. [Google Scholar] [CrossRef]
- Garcillán-Barcia, M.P.; Francia, M.V.; de la Cruz, F. The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol. Rev. 2009, 33, 657–687. [Google Scholar] [CrossRef]
- Zhang, A.; Xu, C.; Wang, H.; Lei, C.; Liu, B.; Guan, Z.; Yang, C.; Yang, Y.; Peng, L. Presence and new genetic environment of pleuromutilin-lincosamide-streptogramin A resistance gene lsa(E) in Erysipelothrix rhusiopathiae of swine origin. Vet. Microbiol. 2015, 177, 162–167. [Google Scholar] [CrossRef]
- Zhou, K.; Zhu, D.; Tao, Y.; Xie, L.; Han, L.; Zhang, Y.; Sun, J. New genetic context of lnu(B) composed of two multi-resistance gene clusters in clinical Streptococcus agalactiae ST-19 strains. Antimicrob. Resist. Infect. Control 2019, 8, 117. [Google Scholar] [CrossRef]
- Wang, M.; Liu, P.; Zhou, Q.; Tao, W.; Sun, Y.; Zeng, Z. Estimating the contribution of bacteriophage to the dissemination of antibiotic resistance genes in pig feces. Environ. Pollut. 2018, 238, 291–298. [Google Scholar] [CrossRef]
- Webster, J.; Bowring, B.; Stroud, L.; Marsh, I.; Sales, N.; Bogema, D. Population Structure and Genomic Characteristics of Australian Erysipelothrix rhusiopathiae Reveals Unobserved Diversity in the Australian Pig Industry. Microorganisms 2023, 11, 297. [Google Scholar] [CrossRef]
- Forde, T.L.; Kollanandi Ratheesh, N.; Harvey, W.T.; Thomson, J.R.; Williamson, S.; Biek, R.; Opriessnig, T. Genomic and Immunogenic Protein Diversity of Erysipelothrix rhusiopathiae Isolated from Pigs in Great Britain: Implications for Vaccine Protection. Front. Microbiol. 2020, 11, 418. [Google Scholar] [CrossRef]
- Kovalchuk, S.; Babii, A. Draft genome sequence data of Erysipelothrix rhusiopathiae vaccine strain VR-2. Data Brief. 2020, 33, 106352. [Google Scholar] [CrossRef]
- CLSI Supplement Vet06. Methods for Antimicrobial Susceptibility Testing of Infrequently Isolated or Fastidious Bacteria Isolated from Animals, 1st ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- Shiraiwa, K.; Ogawa, Y.; Nishikawa, S.; Eguchi, M.; Shimoji, Y. Identification of serovar 1a, 1b, 2, and 5 strains of Erysipelothrix rhusiopathiae by a conventional gel-based PCR. Vet. Microbiol. 2018, 225, 101–104. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Huang, Y.T.; Liu, P.Y.; Shih, P.W. Homopolish: A method for the removal of systematic errors in nanopore sequencing by homologous polishing. Genome Biol. 2021, 22, 95. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.M.; Chooi, Y.H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef] [PubMed]
- Janßen, T.; Voss, M.; Kühl, M.; Semmler, T.; Philipp, H.-C.; Ewers, C. A Combinational Approach of Multilocus Sequence Typing and Other Molecular Typing Methods in Unravelling the Epidemiology of Erysipelothrix Rhusiopathiae Strains from Poultry and Mammals. Vet. Res. 2015, 46, 84. [Google Scholar] [CrossRef] [PubMed]
- Xihui, Z.; Yanlan, L.; Zhiwei, W.; Zheyu, P.; Zhenshu, S.; Cheng, L.; Jianbiao, L.; Shengliang, C.; Lanying, P.; Yubao, L. Antibiotic resistance of Riemerella anatipestifer and comparative analysis of antibiotic-resistance gene detection methods. Poult. Sci. 2023, 102, 102405. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 2021, 76, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Kwok, A.H.; Li, Y.; Jiang, J.; Jiang, P.; Leung, F.C. Complete genome assembly and characterization of an outbreak strain of the causative agent of swine erysipelas—Erysipelothrix rhusiopathiae SY1027. BMC Microbiol. 2014, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Park, S.Y.; Seo, H.W.; Cho, Y.; Choi, S.G.; Seo, S.; Han, W.; Lee, N.K.; Kwon, H.; Han, J.E.; et al. Pathological and Genomic Findings of Erysipelothrix rhusiopathiae Isolated from a Free-Ranging Rough-Toothed Dolphin Steno bredanensis (Cetacea: Delphinidae) Stranded in Korea. Front. Vet. Sci. 2022, 9, 774836. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, K.; Bloodgood, J.; Mullis, M.M.; Deming, A.C.; Colegrove, K.; Kiel Reese, B. Draft Genome Sequences of Erysipelothrix sp. Strains Isolated from Stranded Septic Bottlenose Dolphins in Alabama, USA. Microbiol. Resour. Announc. 2022, 11, e0027322. [Google Scholar] [CrossRef] [PubMed]
- Söderlund, R.; Formenti, N.; Caló, S.; Chiari, M.; Zoric, M.; Alborali, G.L.; Sørensen Dalgaard, T.; Wattrang, E.; Eriksson, H. Comparative genome analysis of Erysipelothrix rhusiopathiae isolated from domestic pigs and wild boars suggests host adaptation and selective pressure from the use of antibiotics. Microb. Genom. 2020, 6, mgen000412. [Google Scholar] [CrossRef] [PubMed]
- Little, S.V.; Hillhouse, A.E.; Lawhon, S.D. Draft Genome Sequence of Erysipelothrix rhusiopathiae, Isolated from a Canine Case of Diskospondylitis. Microbiol. Resour. Announc. 2020, 9, e00592-20. [Google Scholar] [CrossRef] [PubMed]
- Grazziotin, A.L.; Vidal, N.M.; Hoepers, P.G.; Reis, T.F.M.; Mesa, D.; Caron, L.F.; Ingberman, M.; Beirão, B.C.B.; Zuffo, J.P.; Fonseca, B.B. Comparative genomics of a novel clade shed light on the evolution of the genus Erysipelothrix and characterise an emerging species. Sci. Rep. 2021, 11, 3383. [Google Scholar] [CrossRef]
- Eisenberg, T.; Mühldorfer, K.; Erhard, M.; Fawzy, A.; Kehm, S.; Ewers, C.; Semmler, T.; Blom, J.; Lipski, A.; Rau, J.; et al. Erysipelothrix anatis sp. nov., Erysipelothrix aquatica sp. nov. and Erysipelothrix urinaevulpis sp. nov., three novel species of the genus, and emended description of Erysipelothrix. Int. J. Syst. Evol. Microbiol. 2022, 72, 005454. [Google Scholar] [CrossRef] [PubMed]
- Bang, B.H.; Rhee, M.S.; Chang, D.H.; Park, D.S.; Kim, B.C. Erysipelothrix larvae sp. nov., isolated from the larval gut of the rhinoceros beetle, Trypoxylus dichotomus (Coleoptera: Scarabaeidae). Antonie Leeuwenhoek 2015, 107, 443–451. [Google Scholar] [CrossRef]
- Nüesch-Inderbinen, M.; Haussmann, A.; Treier, A.; Zurfluh, K.; Biggel, M.; Stephan, R. Fattening Pigs Are a Reservoir of Florfenicol-Resistant Enterococci Harboring Oxazolidinone Resistance Genes. J. Food Prot. 2022, 85, 740–746. [Google Scholar] [CrossRef]





| Isolate ID (Genome ID) | 1023 (23S00 176-1) | 267 (23S00171-1) | 1012 (23S00175-1) | 584 (23S00173-1) |
|---|---|---|---|---|
| Serotype | 2 | 5 | 2 | 5 |
| Source | Domestic goose | Domestic goose | Domestic goose | Domestic goose |
| Year of isolation | 2021 | 2021 | 2020 | 2020 |
| ENA Acc. No. | ERR12736637 | ERR12736634 | ERR12736636 | ERR12736635 |
| Genome size (bp) | 1,905,426 a; 9362 b | 1,889,255 a | 1,846,905 a | 1,826,410 a |
| Contigs | 2 | 1 | 1 | 1 |
| Genes | 1862 | 1835 | 1805 | 1790 |
| Proteins (CDS) | 1794 | 1767 | 1743 | 1720 |
| tRNAs | 55 | 55 | 55 | 55 |
| GC content (%) | 36 a 29 b | 37 | 36 | 37 |
| ST (MLST) | 4 | 243 (novel) | 242 (novel) | 32 |
| Phenotypic resistance profile (MIC in μg/mL) c | ERY (>32), TYL (2), LIN (>64), CLI (2), TIA (>64), ENR (>16), TET (64), …………………….. STR (>512), SPE (>512), GEN (>512) | ERY (>32), TYL (>32), LIN (>64), CLI (>16), ENR (8), TET (64), …………………… STR (128), SPE (32), GEN (512) | TYL (2), TET (32), ……………………. STR (64), SPE (128), GEN (>512) | ………………….. STR (64), SPE (32), GEN (512) |
| Resistance genes | erm47, lnuB, lsaE, ant(6)-Ia, spw, tetM | ermB, tetM | tetM | none |
| Mutations in gyrA gene | Thr86→Ile86 | Thr86→Lys86 | Thr86 | Thr86 |
| ICE | Tn916-like, ICEEr1023 | Tn916-like | Tn916-like, ICEEr1012 | none |
| Plasmid | 1 | 0 | 0 | 0 |
| bleO | aadD | ant(9)-Ia (aad9) | spc | str | spw | ant(6)-Ia | aph(3′)-III | lnuB | lsaE | ermG | ermT | mefA | msrD | lnuD-like | erm47 | ermB | tetT | tetM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E. rhusiopathiae strains tested in this work n = 4 | 0 | 0 | 0 | 0 | 0 | 25% (1) | 25% (1) | 0 | 25% (1) | 25% (1) | 0 | 0 | 0 | 0 | 0 | 25% (1) | 25% (1) | 0 | 75% (3) |
| E. rhusiopathiae n = 363 * | 0.3% (1) | 0.3% (1) | 0.5% (2) | 0.3% (1) | 0.5% (2) | 3.3% (12) | 3.3% (12) | 1.9% (7) | 3.6% (13) | 3.6% (13) | 0.3% (1) | 0.3% (1) | 0.3% (1) | 0.3% (1) | 1.1% (4) | 0 | 0 | 0.3% (1) | 16% (58) |
| Erysipelothrix sp. n = 13 * | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7.7% (1) | 7.7% (1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dec, M.; Zomer, A.; Webster, J.; Nowak, T.; Stępień-Pyśniak, D.; Urban-Chmiel, R. Integrative and Conjugative Elements and Prophage DNA as Carriers of Resistance Genes in Erysipelothrix rhusiopathiae Strains from Domestic Geese in Poland. Int. J. Mol. Sci. 2024, 25, 4638. https://doi.org/10.3390/ijms25094638
Dec M, Zomer A, Webster J, Nowak T, Stępień-Pyśniak D, Urban-Chmiel R. Integrative and Conjugative Elements and Prophage DNA as Carriers of Resistance Genes in Erysipelothrix rhusiopathiae Strains from Domestic Geese in Poland. International Journal of Molecular Sciences. 2024; 25(9):4638. https://doi.org/10.3390/ijms25094638
Chicago/Turabian StyleDec, Marta, Aldert Zomer, John Webster, Tomasz Nowak, Dagmara Stępień-Pyśniak, and Renata Urban-Chmiel. 2024. "Integrative and Conjugative Elements and Prophage DNA as Carriers of Resistance Genes in Erysipelothrix rhusiopathiae Strains from Domestic Geese in Poland" International Journal of Molecular Sciences 25, no. 9: 4638. https://doi.org/10.3390/ijms25094638
APA StyleDec, M., Zomer, A., Webster, J., Nowak, T., Stępień-Pyśniak, D., & Urban-Chmiel, R. (2024). Integrative and Conjugative Elements and Prophage DNA as Carriers of Resistance Genes in Erysipelothrix rhusiopathiae Strains from Domestic Geese in Poland. International Journal of Molecular Sciences, 25(9), 4638. https://doi.org/10.3390/ijms25094638