Centuries-Old DNA from an Extinct Population of Aesculapian Snake (Zamenis longissimus) Offers New Phylogeographic Insight


Abstract
:1. Introduction
2. Materials and Methods
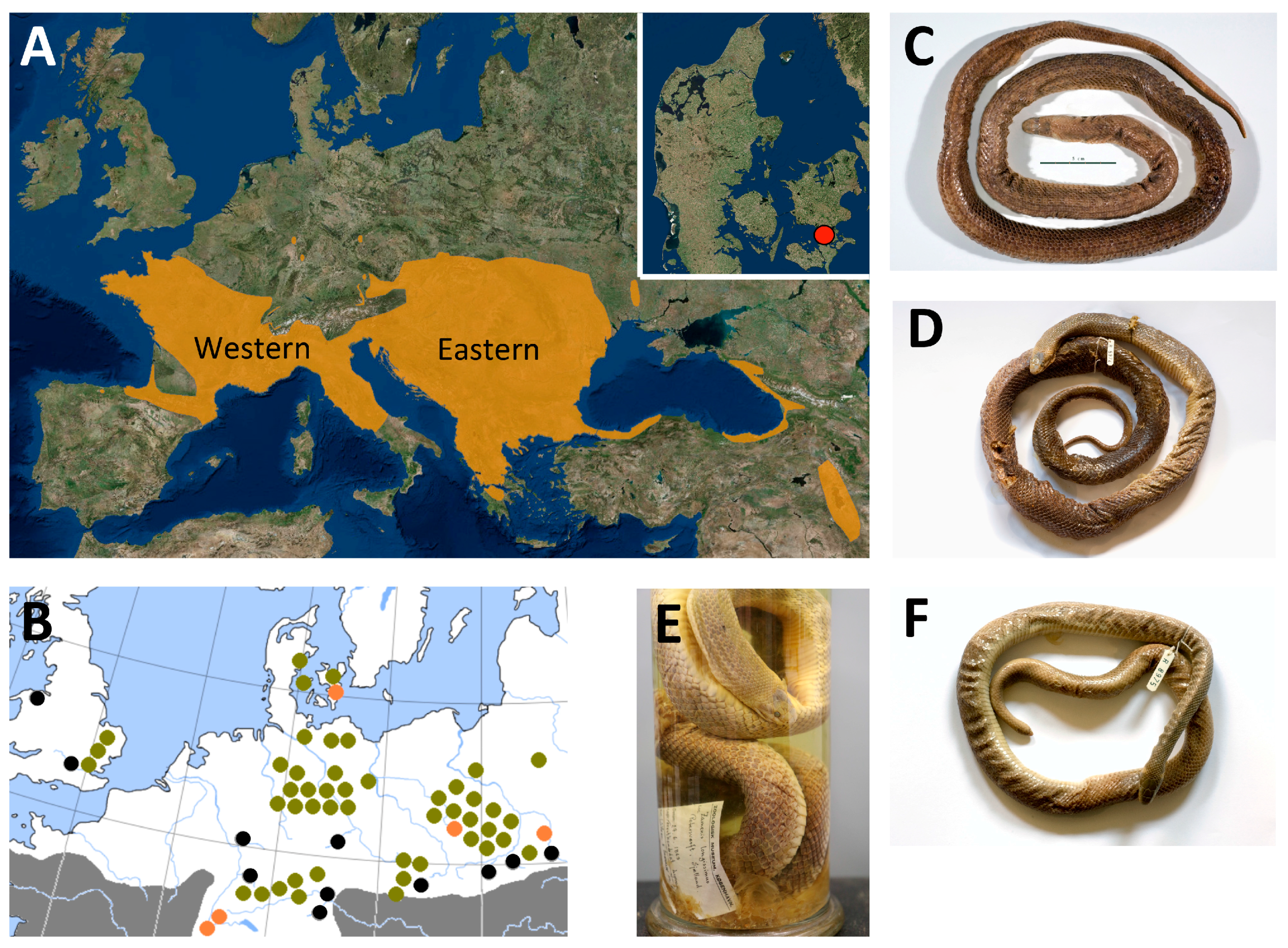
2.1. Samples
2.2. DNA Extraction
2.3. Library Preparation and Sequencing
2.4. Bioinformatics
2.5. Alignment and Network Analysis
3. Results
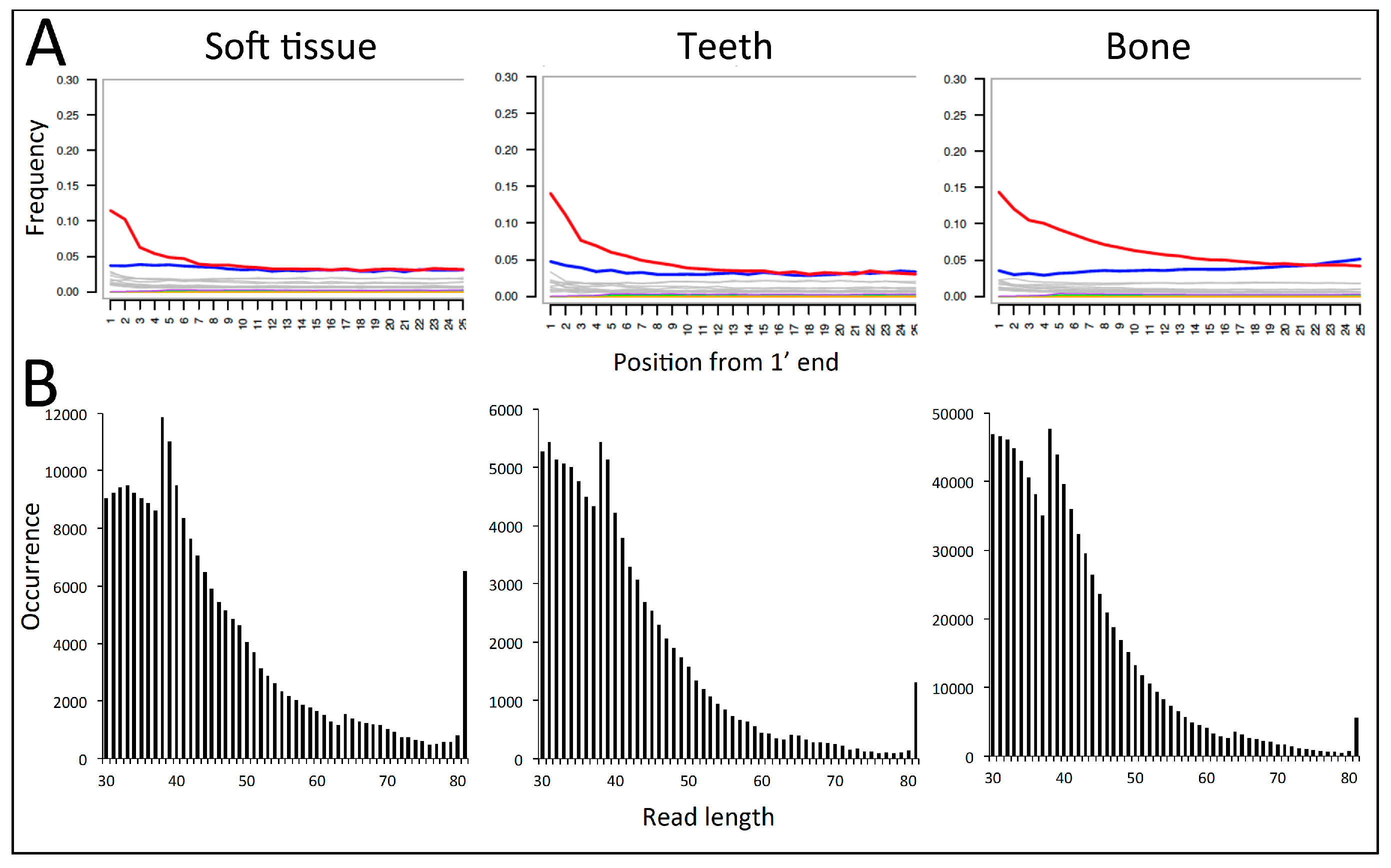
3.1. Sequencing, Trimming, and Mapping
3.2. DNA Preservation
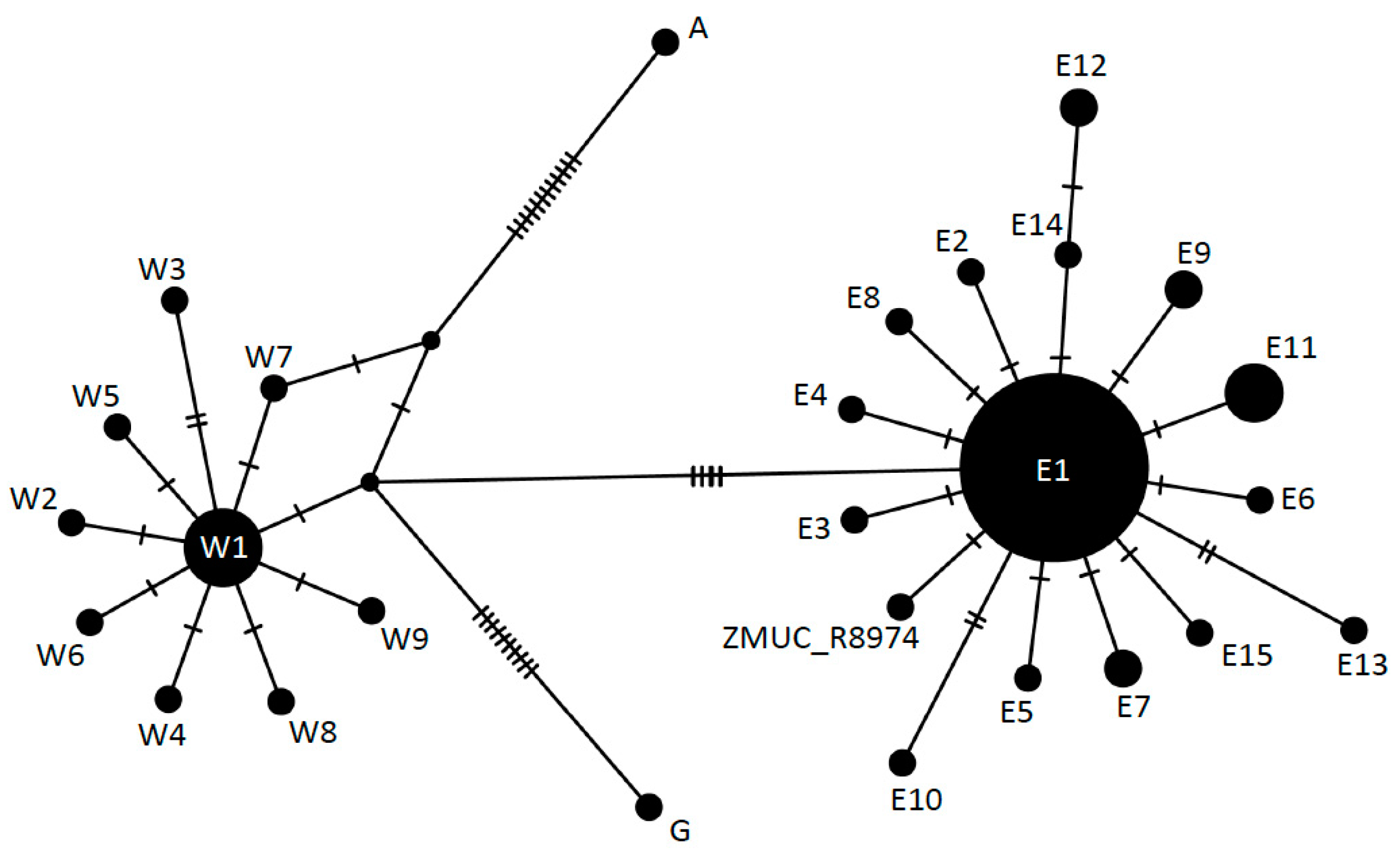
3.3. Phylogeography
4. Discussion
4.1. DNA Preservation
4.2. Phylogeography
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guschanski, K.; Krause, J.; Sawyer, S.; Valente, L.M.; Bailey, S.; Finstermeier, K.; Sabin, R.; Gilissen, E.; Sonet, G.; Nagy, Z.T. Next-generation museomics disentangles one of the largest primate radiations. Syst. Boil. 2013, 62, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Wandeler, P.; Hoeck, P.E.; Keller, L.F. Back to the future: Museum specimens in population genetics. Trends Ecol. Evol. 2007, 22, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heimes, P. Die reptilien des rheingautaunus unter berücksichtigung der schutzproblematik der äskulapnatter, Elaphe longissima (laurenti, 1768). Naturschutz-Zentrum Hessen, Wetzlar 1988. Unpublished work. [Google Scholar]
- Musilová, R.; Zavadil, V.; Kotlík, P. Isolated populations of Zamenis longissimus (reptilia: Squamata) above the northern limit of the continuous range in europe: Origin and conservation status. Acta Soc. Zool. Bohem. 2007, 71, 197–208. [Google Scholar]
- Waitzmann, M. Zur situation der äskulapnatter Elaphe longissima (laurenti, 1768) in der bundesrepublik deutschland. Mertensiella 1993, 3, 115–133. [Google Scholar]
- Gomille, A. Die Äskulapnatter-Elaphe longissima: Verbreitung und Lebensweise in Mitteleuropa; Edition Chimaira: Frankfurt am Main, Germany, 2002; pp. 1–158. [Google Scholar]
- Szyndlar, Z. Fossil snakes from poland. Acta Zool. Cracov. 1984, 28, 1–156. [Google Scholar]
- Szyndlar, Z. A review of neogene and quaternary snakes of central and eastern europe. Part i: Scolecophidia, Boidae, Colubrinae. Estudios Geológicos 1991, 47, 103–126. [Google Scholar]
- Ljungar, L. First subfossil find of the aesculapian snake. Amphibia-Reptilia 1995, 16, 93–94. [Google Scholar] [CrossRef]
- Sommer, R.S.; Persson, A.; Wieseke, N.; Fritz, U. Holocene recolonization and extinction of the pond turtle, Emys orbicularis (l., 1758), in europe. Quat. Sci. Rev. 2007, 26, 3099–3107. [Google Scholar] [CrossRef]
- Richter, J.; Noe-Nygaard, N. A late mesolithic hunting station at agernæs, fyn, denmark. Acta Archaeol. 2003, 74, 1–64. [Google Scholar] [CrossRef]
- Kristensen, H.V. Første danske fund af knogler fra æskulapsnog (Zamenis longissimus) i bronzealdergrav. Nord. Herpetol. Foren. 2008, 51, 82–86. [Google Scholar]
- Boulenger, G.A. The Snakes of Europe; Methuen & Company, Limited: London, UK, 1913. [Google Scholar]
- Sarauw, G. En stenalders boplads i maglemose ved mullerup sammenholdt med beslaegtede. In Aarbøger for Nordisk Oldkyndighed; Lund Universitet: Lund, Sweden, 1903; Volume 1903, pp. 148–315. [Google Scholar]
- Sarauw, G.F. Rodsymbiose og Mykorrhizer Saerlig Hos Skovtraeerne; H. Hagerups Forlag: Copenhagen, Denmark, 1893; Volume 18, pp. 127–259. [Google Scholar]
- Fog, K.; Schmedes, A.; de Lasson, D.R. Nordens Padder og Krybdyr; G.E. C. Gad: Copenhagen, Denmark, 1997. [Google Scholar]
- Hvass, H. Danmarks æskulapsnoge [Danish aesculapian snake]. Naturens Verden 1942, 26, 74–86. [Google Scholar]
- Munk, H. Hassselskoven. En Skov- og Landbrugskulturhistorisk Studie fra Sydsjælland i 1600–1700; Holger Munk: Frederikssund, Denmark, 1969; p. 249. [Google Scholar]
- Musilová, R.; Zavadil, V.; Marková, S.; Kotlík, P. Relics of the europe’s warm past: Phylogeography of the aesculapian snake. Mol. Phylogenet. Evol. 2010, 57, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Allentoft, M.E.; Sikora, M.; Sjögren, K.-G.; Rasmussen, S.; Rasmussen, M.; Stenderup, J.; Damgaard, P.B.; Schroeder, H.; Ahlström, T.; Vinner, L. Population genomics of bronze age eurasia. Nature 2015, 522, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Willerslev, E.; Cooper, A. Ancient DNA. Proc. R. Soc. Lond. B Biol. Sci. 2005, 272, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Orlando, L.; Ginolhac, A.; Zhang, G.; Froese, D.; Albrechtsen, A.; Stiller, M.; Schubert, M.; Cappellini, E.; Petersen, B.; Moltke, I.; et al. Recalibrating equus evolution using the genome sequence of an early middle pleistocene horse. Nature 2013, 499, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protoc. 2010, 2010, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Lindgreen, S.; Orlando, L. Adapterremoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.C.; Wilson, R. Thamnophis Sirtalis Isolate EDBJR-23777, Whole Genome Shotgun Sequencing Project. 2015. Available online: https://www.ncbi.nlm.nih.gov/nuccore/LFLD00000000.1. (accessed on 9 February 2018).
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Allentoft, M.E.; Collins, M.; Harker, D.; Haile, J.; Oskam, C.L.; Hale, M.L.; Campos, P.F.; Samaniego, J.A.; Gilbert, M.T.P.; Willerslev, E. The half-life of DNA in bone: Measuring decay kinetics in 158 dated fossils. Proc. R. Soc. Lond. B Biol. Sci. 2012, 279, 4724–4733. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.R.; Elmberg, J.; Sanders, K.L.; Gravlund, P. Rediscovery of the rare sea snake Hydrophis parviceps smith 1935: Identification and conservation status. Copeia 2012, 2012, 276–282. [Google Scholar] [CrossRef]
- Friedman, M.; DeSalle, R. Mitochondrial DNA extraction and sequencing of formalin-fixed archival snake tissue. DNA Seq. 2008, 19, 433–437. [Google Scholar] [CrossRef]
- Ruane, S.; Austin, C.C. Phylogenomics using formalin-fixed and 100+ year-old intractable natural history specimens. Mol. Ecol. Resour. 2017, 17, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.; Rasmussen, A.R.; Simonsen, K.P. Enzymatic detection of formalin-fixed museum specimens for DNA analysis and enzymatic maceration of formalin-fixed specimens. Collect. Forum 2016, 30, 1–6. [Google Scholar] [CrossRef]
- Joger, U.; Fritz, U.; Guicking, D.; Kalyabina-Hauf, S.; Nagy, Z.T.; Wink, M. Phylogeography of western palaearctic reptiles–spatial and temporal speciation patterns. Zool. Anz. 2007, 246, 293–313. [Google Scholar] [CrossRef]
- Joger, U.; Fritz, U.; Guicking, D.; Kalyabina-Hauf, S.; Nagy, Z.T.; Wink, M. Relict populations and endemic clades in palearctic reptiles: Evolutionary history and implications for conservation. In Relict Species; Springer: Berlin, Germany, 2010; pp. 119–143. [Google Scholar]
- Boehme, M.U.; Fritz, U.; Kotenko, T.; Ljubisavljević, K.; Tzankov, N.; Berendonk, T.U. Phylogeography and cryptic variation within the Lacerta viridis complex (lacertidae, reptilia). Zool. Scr. 2007, 36, 119–131. [Google Scholar] [CrossRef]
- Guicking, D.; Lawson, R.; Joger, U.; Wink, M. Evolution and phylogeny of the genus Natrix (serpentes: Colubridae). Biol. J. Linn. Soc. 2006, 87, 127–143. [Google Scholar] [CrossRef]
- Lenk, P.; Fritz, U.; Joger, U.; Wink, M. Mitochondrial phylogeography of the european pond turtle, Emys orbicularis (linnaeus 1758). Mol. Ecol. 1999, 8, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Sommer, R.S.; Fritz, U.; Seppä, H.; Ekström, J.; Persson, A.; Liljegren, R. When the pond turtle followed the reindeer: Effect of the last extreme global warming event on the timing of faunal change in northern europe. Glob. Chang. Biol. 2011, 17, 2049–2053. [Google Scholar] [CrossRef]
- Kindler, C.; Graciá, E.; Fritz, U. Extra-mediterranean glacial refuges in barred and common grass snakes (Natrix helvetica, N. natrix). Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Böhme, W. Äskulapnatter (elaphe longissima laurenti 1768). Handbuch der Reptilien und Amphibien Europas 1993, 3, 331–372. [Google Scholar]
- Mikátová, B.; Zavadil, V. Aesculapian snake—Elaphe longissima (laurenti, 1768). In Atlas of the Distribution of Reptiles in the Czech Republic; Mikátová, B., Vlašín, M., Zavadil, V., Eds.; Agentura Ochrany Prirody: Brno-Prague, Czech Republic, 2001; pp. 113–123. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Sample | Substrate | Total | Retained | Mapped | Non-Clonal | Hits, % | Efficiency, % | C-T, % | Av. Length, bp |
|---|---|---|---|---|---|---|---|---|---|
| HK2489 | muscle/skin | 172,107,931 | 147,491,661 | 8,696,705 | 884,972 | 5.9 | 0.51 | 15.5 | 47.1 |
| ZMUC R8974 | muscle/skin | 76,664,518 | 63,973,314 | 5,497,285 | 365,276 | 8.6 | 0.48 | 11.0 | 48.6 |
| ZMUC R8974 | teeth | 59,814,253 | 53,875,108 | 591,382 | 149,579 | 1.1 | 0.25 | 13.8 | 44.2 |
| ZMUC R8974 | vertebra | 108,710,374 | 91,998,453 | 4,480,788 | 1,416,148 | 4.9 | 1.30 | 14.7 | 43.1 |
| ZMUC R8975 | muscle/skin | 33,202,047 | 22,458,083 | 139,615 | 18,940 | 0.6 | 0.06 | 11.2 | 45.7 |
| ZMUC R8975 | teeth | 50,978,945 | 40,947,311 | 453,775 | 73,277 | 1.1 | 0.14 | 15.2 | 42.0 |
| Number of Reads Matching | |||||
|---|---|---|---|---|---|
| Sample | Eastern | Western | Greek | Asian | Z. lineatus |
| HK2489 | 467 | 463 | 460 | 453 | 169 |
| ZMUC R8974 | 2225 | 2197 | 2131 | 2088 | 718 |
| ZMUC R8975 | 4 | 4 | 4 | 4 | 3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allentoft, M.E.; Rasmussen, A.R.; Kristensen, H.V. Centuries-Old DNA from an Extinct Population of Aesculapian Snake (Zamenis longissimus) Offers New Phylogeographic Insight. Diversity 2018, 10, 14. https://doi.org/10.3390/d10010014
Allentoft ME, Rasmussen AR, Kristensen HV. Centuries-Old DNA from an Extinct Population of Aesculapian Snake (Zamenis longissimus) Offers New Phylogeographic Insight. Diversity. 2018; 10(1):14. https://doi.org/10.3390/d10010014
Chicago/Turabian StyleAllentoft, Morten E., Arne Redsted Rasmussen, and Hans Viborg Kristensen. 2018. "Centuries-Old DNA from an Extinct Population of Aesculapian Snake (Zamenis longissimus) Offers New Phylogeographic Insight" Diversity 10, no. 1: 14. https://doi.org/10.3390/d10010014
APA StyleAllentoft, M. E., Rasmussen, A. R., & Kristensen, H. V. (2018). Centuries-Old DNA from an Extinct Population of Aesculapian Snake (Zamenis longissimus) Offers New Phylogeographic Insight. Diversity, 10(1), 14. https://doi.org/10.3390/d10010014