
Full-Length Transcriptomics Reveal the Gene Expression Profiles of Reef-Building Coral Pocillopora damicornis and Symbiont Zooxanthellae


Abstract
:
1. Introduction
2. Methods and Materials
2.1. Ethics
2.2. Sample Collection
2.3. RNA Extraction
2.4. Library Construction and Sequencing
2.5. Data Processing
2.6. Error Correction Using Illumina Reads
2.7. Removing Redundancies
2.8. Gene Functional Annotation
2.9. CDS Prediction
2.10. TF Analysis
2.11. SSR Analysis
2.12. LncRNA Analysis
2.13. Quantification of Gene Expression Levels
2.14. Correlation Analysis of Gene Expression
2.15. GO and KEGG Enrichment Analysis
3. Results
3.1. Raw Data Quality Control
3.2. Transcript Correction
3.3. Redundant Removal
3.4. Gene Function Annotation
3.4.1. NR Database Annotation
3.4.2. GO Classification
3.4.3. KOG Classification
3.4.4. KEGG Classification
3.4.5. Pfam Database Annotation
3.4.6. Swiss-Prot Database Comment
3.5. Gene Structure Analysis
3.5.1. CDS Prediction
3.5.2. TF Analysis
3.5.3. SSR Analysis
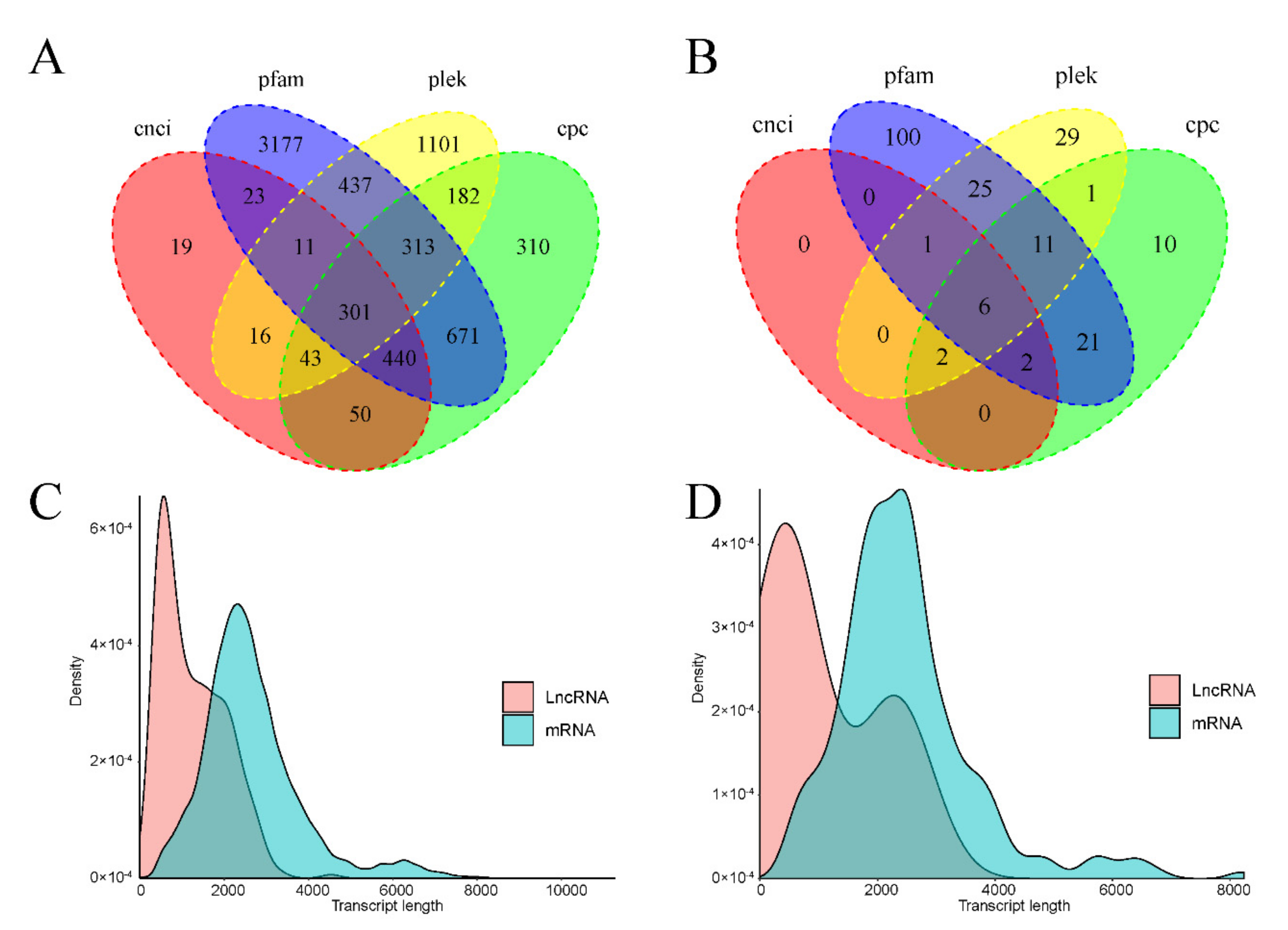
3.5.4. LncRNA Prediction
3.6. Gene Expression Analysis
3.6.1. Reference Sequence Alignment
3.6.2. Gene Expression Statistics
3.7. Enrichment Analysis of Coral High-Expression Genes
3.7.1. GO Entries
3.7.2. KEGG Entries
3.8. Functional Gene Analysis of Zooxanthellae
4. Discussion
4.1. Coral Biological Process
4.2. Coral Cellular Component
4.3. Coral Molecular Function
4.4. Coral Mineral Absorption
4.5. Gene Function of Zooxanthellae
4.6. Biochemical Connection in the Coral and Algal Symbiont
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Muscatine, L. Glycerol Excretion by Symbiotic Algae from Corals and Tridacna and Its Control by the Host. Science 1967, 156, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Odum, H.T.; Odum, E.P. Trophic Structure and Productivity of a Windward Coral Reef Community on Eniwetok Atoll. Ecol. Monogr. 1955, 25, 291–320. [Google Scholar] [CrossRef]
- Costanza, R.; De Groot, R.; Sutton, P.; Van der Ploeg, S.; Anderson, S.J.; Kubiszewski, I.; Farber, S.; Turner, R.K. Changes in the global value of ecosystem services. Glob. Environ. Chang. 2014, 26, 152–158. [Google Scholar] [CrossRef]
- Moberg, F.; Folke, C. Ecological goods and services of coral reef ecosystems. Ecol. Econ. 1999, 29, 215–233. [Google Scholar] [CrossRef]
- Reimer, J.D.; Kise, H.; Wee, H.B.; Lee, C.L.; Soong, K. Crown-of-thorns starfish outbreak at oceanic Dongsha Atoll in the northern South China Sea. Mar. Biodivers. 2019, 49, 2495–2497. [Google Scholar] [CrossRef]
- Foster, N.L.; Attrill, M.J. Changes in coral reef ecosystems as an indication of climate and global change. In Climate Change, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 427–443. [Google Scholar]
- Kleypas, J.A.; Buddemeier, R.W.; Gattuso, J.P. The future of coral reefs in an age of global change. Int. J. Earth Sci. 2001, 90, 426–437. [Google Scholar] [CrossRef]
- Benhaim, Y.; Zichermankeren, M.; Rosenberg, E. Temperature-Regulated Bleaching and Lysis of the Coral Pocillopora damicornis by the Novel Pathogen Vibrio coralliilyticus. Appl. Environ. Microbiol. 2003, 69, 4236. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, E.; Ben-Haim, Y. Microbial diseases of corals and global warming. Environ. Microbiol. 2002, 4, 318–326. [Google Scholar] [CrossRef]
- Zhou, G.; Tong, H.; Cai, L.; Huang, H. Transgenerational Effects on the Coral Pocillopora damicornis Microbiome under Ocean Acidification. Microb. Ecol. 2021, 68, 1–9. [Google Scholar]
- Camaya, A.P. Stages of the symbiotic zooxanthellae-host cell division and the dynamic role of coral nucleus in the partitioning process: A novel observation elucidated by electron microscopy. Coral Reefs 2020, 39, 929–938. [Google Scholar] [CrossRef]
- Huang, C.; Leng, D.; Sun, S.; Zhang, X.D. Re-analysis of the coral Acropora digitifera transcriptome reveals a complex lncRNAs-mRNAs interaction network implicated in Symbiodinium infection. BMC Genom. 2019, 20, 48. [Google Scholar] [CrossRef] [Green Version]
- Brown, B. Coral bleaching: Causes and consequences. Coral Reefs 1997, 16, S129–S138. [Google Scholar] [CrossRef]
- Muscatine, L.; Porter, J.W. Reef corals—Mutualistic symbioses adapted to nutrient-poor environments. Bioscience 1977, 27, 454–460. [Google Scholar] [CrossRef]
- Kuzminov, F.I.; Brown, C.M.; Fadeev, V.V.; Gorbunov, M.Y. Effects of metal toxicity on photosynthetic processes in coral symbionts, Symbiodinium spp. J. Exp. Mar. Biol. Ecol. 2013, 446, 216–227. [Google Scholar] [CrossRef]
- Rowan, R.; Knowlton, N.; Baker, A.; Jara, J.H. Landscape ecology of algal symbionts creates variation in episodes of coral bleaching. Nature 1997, 388, 265–269. [Google Scholar] [CrossRef]
- LaJeunesse, T. Diversity and community structure of symbiotic dinoflagellates from Caribbean coral reefs. Mar. Biol. 2002, 141, 387–400. [Google Scholar]
- Al-Sofyani, A.A.; Floos, Y. Effect of temperature on two reef-building corals Pocillopora damicornis and P. verrucosa in the red sea. Oceanologia 2013, 55, 917–935. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, D.A.; Petrou, K.; Gates, R.D. Coral bleaching from a single cell perspective. ISME J. 2018, 12, 1558–1567. [Google Scholar] [CrossRef]
- Jiang, L.; Guo, M.-L.; Zhang, F.; Zhang, Y.-Y.; Zhou, G.-W.; Lei, X.-M.; Yuan, X.-C.; Sun, Y.-F.; Yuan, T.; Cai, L.; et al. Impacts of elevated temperature and pCO2 on the brooded larvae of Pocillopora damicornis from Luhuitou reef, China: Evidence for local acclimatization. Coral Reefs 2020, 39, 331–344. [Google Scholar] [CrossRef]
- Rosado, P.M.; Leite, D.C.D.A.; Duarte, G.A.S.; Chaloub, R.M.; Jospin, G.; da Rocha, U.N.; Saraiva, J.P.; Dini-Andreote, F.; Eisen, J.A.; Bourne, D.G.; et al. Marine probiotics: Increasing coral resistance to bleaching through microbiome manipulation. ISME J. 2019, 13, 921–936. [Google Scholar] [CrossRef] [Green Version]
- LaJeunesse, T.C. Zooxanthellae. Curr. Biol. 2020, 30, R1110–R1113. [Google Scholar] [CrossRef]
- Vidal-Dupiol, J.; Zoccola, D.; Tambutté, E.; Grunau, C.; Cosseau, C.; Smith, K.M.; Freitag, M.; Dheilly, N.M.; Allemand, D.; Tambutté, S. Genes related to ion-transport and energy production are upregulated in response to CO2-driven pH decrease in corals: New insights from transcriptome analysis. PLoS ONE 2013, 8, e58652. [Google Scholar] [CrossRef] [Green Version]
- Sampayo, E.M.; Franceschinis, L.; Hoegh-Guldberg, O.; Dove, S. Niche partitioning of closely related symbiotic dinoflagellates. Mol. Ecol. 2010, 16, 3721–3733. [Google Scholar] [CrossRef]
- Zhou, Z.; Yu, X.; Tang, J.; Zhu, Y.; Chen, G.; Guo, L.; Huang, B. Dual recognition activity of a rhamnose-binding lectin to pathogenic bacteria and zooxanthellae in stony coral Pocillopora damicornis. Dev. Comp. Immunol. 2017, 70, 88–93. [Google Scholar] [CrossRef]
- Tombácz, D.; Sharon, D.; Oláh, P.; Csabai, Z.; Snyder, M.; Boldogkői, Z. Strain Kaplan of Pseudorabies Virus Genome Sequenced by PacBio Single-Molecule Real-Time Sequencing Technology. Genome Announc. 2014, 2, e00628-14. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Korlach, J. Nonhybrid, finished microbial genome assemblies from long-read smrt sequencing data. Nat. Methods 2013, 10, 563. [Google Scholar] [CrossRef]
- Chen, S.; Qiu, G.; Yang, M. SMRT sequencing of full-length transcriptome of seagrasses Zostera japonica. Sci. Rep. 2019, 9, 14537. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION sequencing and genome assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.-S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; Omalley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Cherry, J.M. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Shimizu, K.; Adachi, J.; Muraoka, Y. ANGLE: A sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinform. Comput. Biol. 2006, 4, 649–664. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.; Giovannoni, J.J.; et al. ITAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Molecular Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-M.; Liu, T.; Liu, C.-J.; Song, S.; Zhang, X.; Liu, W.; Jia, H.; Xue, Y.; Guo, A.-Y. Animaltfdb 2.0: A resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2015, 1, D76. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing Sequence Intrinsic Composition to Classify Protein-Coding and Long Non-Coding Transcripts. Nucleic Acids Res. 2013, 17, e166. [Google Scholar] [CrossRef]
- Aimin, L.; Junying, Z.; Zhongyin, Z. PLEK: A Tool for Predicting Long Non-Coding Rnas and Messenger Rnas Based on an Improved K-Mer Scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Boyer, P.D. The atp synthase—A splendid molecular machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef] [Green Version]
- Stock, D.; Leslie, A.G.; Walker, J.E. Molecular architecture of the rotary motor in atp synthase. Science 1999, 286, 1700–1705. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, A.; Mesnildrey, S.; Xu, Y.; Moréra, S.; Janin, J.; Véron, M. Nucleoside diphosphate kinase. J. Biol. Chem. 1996, 271, 19928–19934. [Google Scholar] [CrossRef] [Green Version]
- Kurakawa, T.; Ueda, N.; Maekawa, M.; Kobayashi, K.; Kojima, M.; Nagato, Y.; Sakakibara, H.; Kyozuka, J. Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 2007, 445, 652–655. [Google Scholar] [CrossRef]
- Ramakrishnan, V. Ribosome structure and the mechanism of translation. Cell 2002, 108, 557–572. [Google Scholar] [CrossRef] [Green Version]
- Jamin, A.; Wiebe, M.S. Barrier to autointegration factor (banf1): Interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr. Opin. Cell Biol. 2015, 34, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Nyberg, J.; Csapó, J.; Malmgren, B.A. Changes in the d- and l-content of aspartic acid, glutamic acid, and alanine in a scleractinian coral over the last 300 years. Org. Geochem. 2001, 32, 623–632. [Google Scholar] [CrossRef]
- Hsu, S.F.; Lai, H.C.; Jinn, T.L. Cytosol-localized heat shock factor-binding protein, athsbp, functions as a negative regulator of heat shock response by translocation to the nucleus and is required for seed development in arabidopsis. Plant Physiol. 2010, 153, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, H.; Shinoda, T.; Cornelius, F.; Toyoshima, C. Crystal structure of the sodium-potassium pump (na+, k+-atpase) with bound potassium and ouabain. Proc. Natl. Acad. Sci. USA 2009, 106, 13742–13747. [Google Scholar] [CrossRef] [Green Version]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 589–599. [Google Scholar] [CrossRef]
- Bennett, V.; Otto, E.; Davis, J.; Davis, L.; Kordeli, E. Chapter 5 ankyrins: A family of proteins that link diverse membrane proteins to the spectrin skeleton. Curr. Top. Membr. 1991, 38, 65–77. [Google Scholar]
- Kanter, U.; Usadel, B.; Guerineau, F.; Yong, L.; Pauly, M.; Tenhaken, R. The inositol oxygenase gene family of arabidopsis is involved in the biosynthesis of nucleotide sugar precursors for cell-wall matrix polysaccharides. Planta 2005, 221, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Ludewig, U.; von Wirén, N.; Frommer, W.B. Uniport of nh by the root hair plasma membrane Ammonium Transporter LeAMT1;1. J. Biol. Chem. 2002, 277, 13548–13555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizutani, K.; Ito, H.; Iwamoto, I.; Morishita, R.; Deguchi, T.; Nozawa, Y.; Asano, T.; Nagata, K.-I. Essential roles of erk-mediated phosphorylation of vinexin in cell spreading, migration and anchorage-independent growth. Oncogene 2007, 26, 7122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, G.; Smith, M.J.; Macer, D.; Webster, P.; Mortara, R.A. Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. J. Cell Sci. 1986, 86, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, G.; Valtorta, F.; Torielli, L.; Chieregatti, E.; Bianchi, G. Hypertension-associated point mutations in the adducin alpha and beta subunits affect actin cytoskeleton and ion transport. J. Clin. Investig. 1996, 97, 2815–2822. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, H. Plant development: Regulation by protein degradation. Science 2002, 297, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Dumoulin, B.; Ufer, C.; Stehling, S.; Heydeck, D.; Kuhn, H.; Sofi, S. Identification of the COMM-domain containing protein 1 as specific binding partner for the guanine-rich RNA sequence binding factor 1. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129678. [Google Scholar] [CrossRef]
- Weng, M.-T.; Tung, T.-H.; Lee, J.-H.; Wei, S.-C.; Lin, H.-L.; Huang, Y.-J.; Wong, J.-M.; Luo, J.; Sheu, J.-C. Enhancer of rudimentary homolog regulates DNA damage response in hepatocellular carcinoma. Sci. Rep. 2015, 5, 9357. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Y.; Zhang, H.; Zhai, H.; Liu, Q.; He, S. A soluble starch synthase I gene, IbSSI, alters the content, composition, granule size and structure of starch in transgenic sweet potato. Sci. Rep. 2017, 7, 2315. [Google Scholar] [CrossRef] [Green Version]
- Tijet, N.; Brash, A.R. Allene oxide synthases and allene oxides. Prostaglandins Other Lipid Mediat. 2002, 68, 423–431. [Google Scholar] [CrossRef]
- Kim, C.W.; Goldberger, O.A.; Gallo, R.L.; Bernfield, M. Members of the syndecan family of heparan sulfate proteoglycans are expressed in distinct cell-, tissue-, and development-specific patterns. Mol. Biol. Cell 1994, 5, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Gruez, A.; Pignol, D.; Zeghouf, M.; Covès, J.; Fontecave, M.; Ferrer, J.-L.; Fontecilla-Camps, J.C. Four crystal structures of the 60 kda flavoprotein monomer of the sulfite reductase indicate a disordered flavodoxin-like module. J. Mol. Biol. 2000, 299, 199–212. [Google Scholar] [CrossRef]
- Salman, Z.K.; Refaat, R.; Selima, E.; Sarha, A.E.; Ismail, M.A. The combined effect of metformin and l-cysteine on inflammation, oxidative stress and insulin resistance in streptozotocin-induced type 2 diabetes in rats. Eur. J. Pharmacol. 2013, 714, 448–455. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, C.; Jiang, C.; Pan, J.; Zhang, G.; Liu, H.; Zhang, H. The tumor necrosis factor receptor-associated factor (TRAF)-like family protein SEVEN IN ABSENTIA 2 (SINA2) promotes drought tolerance in an ABA-dependent manner in Arabidopsis. New Phytol. 2014, 202, 174–187. [Google Scholar] [CrossRef]
- Pedro, F.J.; Xinnian, D.; Devarenne, T.P. Functional characterization of a nudix hydrolase atnudx8 upon pathogen attack indicates a positive role in plant immune responses. PLoS ONE 2014, 9, e114119. [Google Scholar]
- Jing, J.; Hao, Z.; Kang, Y.; Bina, D.; Lo, C.S.; Blankenship, R.E. Characterization of the peridinin–chlorophyll a-protein complex in the dinoflagellate Symbiodinium. Biochim. Biophys. Acta 2012, 1817, 983–989. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.C.; Somanchi, A.; Mayfield, S.P. Interorganellar crosstalk: New perspectives on signaling from the chloroplast to the nucleus. Genome Biol. 2001, 2, 1–4. [Google Scholar]
- Bollenbach, T.J.; Sharwood, R.; Gutierrez, R.; Lerbs-Mache, S.; Stern, D.B. The RNA-binding proteins CSP41a and CSP41b may regulate transcription and translation of chloroplast-encoded RNAs in Arabidopsis. Plant Mol. Biol. 2009, 69, 541–552. [Google Scholar] [CrossRef]
- Meyer, T.E. Evolution of photosynthetic reaction centers and light harvesting chlorophyll proteins. Biosystems 1994, 33, 167–175. [Google Scholar] [CrossRef]
- Alves-Filho, J.C.; Pålsson-McDermott, E.M. Pyruvate Kinase M2: A Potential Target for Regulating Inflammation. Front. Immunol. 2016, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Minges, A.; Groth, G. Small-molecule inhibition of pyruvate phosphate dikinase targeting the nucleotide binding site. PLoS ONE 2017, 12, e0181139. [Google Scholar] [CrossRef] [Green Version]
- Kai, Y.; Matsumura, H.; Izui, K. Phosphoenolpyruvate carboxylase: Three-dimensional structure and molecular mechanisms. Arch. Biochem. Biophys. 2013, 414, 170–179. [Google Scholar] [CrossRef]
- Kodaimati Mohamad, S.; Lian, S.; Schatz George, C.; Weiss Emily, A. Energy transfer-enhanced photocatalytic reduction of protons within quantum dot light-harvesting–catalyst assemblies. Proc. Natl. Acad. Sci. USA 2018, 115, 201805625. [Google Scholar] [CrossRef] [Green Version]
- Suggett, D.J.; Warner, M.E.; Leggat, W. Symbiotic Dinoflagellate Functional Diversity Mediates Coral Survival under Ecological Crisis. Trends Ecol. Evol. 2017, 32, 735–745. [Google Scholar] [CrossRef]
- Zhang, Y.; Ip, J.C.; Xie, J.Y.; Yeung, Y.H.; Sun, Y.; Qiu, J.W. Host-symbiont transcriptomic changes during natural bleaching and recovery in the leaf coral Pavona decussata. Sci. Total Environ. 2021, 806, 150656. [Google Scholar]
- Gates, R.D.; Hoegh-Guldberg, O.; McFall-Ngai, M.J.; Bil, K.Y.; Muscatine, L. Free amino acids exhibit anthozoan “host factor” activity: They induce the release of photosynthate from symbiotic dinoflagellates in vitro. Proc. Natl. Acad. Sci. USA 1995, 92, 7430–7434. [Google Scholar] [CrossRef] [Green Version]
- Gates, R.D.; Bil, K.Y.; Muscatine, L. The influence of an anthozoan “host factor” on the physiology of a symbiotic dinoflagellate. J. Exp. Mar. Biol. Ecol. 1999, 232, 241–259. [Google Scholar] [CrossRef]
- Cook, C.B.; Davy, S.K. Are free amino acids responsible for the ‘host factor’ effects on symbiotic zooxanthellae in extracts of host tissue? Hydrobiologia 2001, 461, 71–78. [Google Scholar] [CrossRef]
- Muscatine, L.; Karakashian, S.J.; Karakashian, M.W. Soluble Extracellular Products of Algae Symbiotic with a Ciliate, a Sponge and a Mutant Hydra. Comp. Biochem. Physiol. 1967, 20, 1–6. [Google Scholar] [CrossRef]
- Muscatine, L.; Hand, C. Direct Evidence for the Transfer of Materials from Symbiotic Algae to the Tissues of a Coelenterate. Proc. Natl. Acad. Sci. USA 1958, 44, 1259–1263. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-J.; Huang, Z.-Y.; Lin, C.-Y.; Wang, L.-H.; Chou, P.-H.; Chen, C.-S.; Li, H.-H. Generation of clade- and symbiont-specific antibodies to characterize marker molecules during Cnidaria-Symbiodinium endosymbiosis. Sci. Rep. 2017, 7, 5488. [Google Scholar] [CrossRef]
- Jiang, P.L.; Pasaribu, B.; Chen, C.S. Nitrogen-deprivation elevates lipid levels in Symbiodinium spp. by lipid droplet accumulation: Morphological and compositional analyses. PLoS ONE 2014, 9, e87416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goreau, T.F.; Goreau, N.I.; Yonge, C.M. On the utilization of photosynthetic products from zooxanthellae and of a dissolved amino acid in Tridacna maxima f. elongata (Mollusca: Bivalvia). J. Zool. 2010, 169, 417–454. [Google Scholar] [CrossRef]
- Trench, R.K. The Physiology and Biochemistry of Zooxanthellae Symbiotic with Marine Coelenterates. II. Liberation of Fixed 14C by Zooxanthellae in Vitro. Proc. R. Soc. Lond. B Biol. Sci. 1971, 177, 237–250. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistical Items | Results |
|---|---|
| CCS number | 602,185 |
| CCS mean num passes | 8 |
| NFL | 116,774 |
| FL | 485,411 |
| FLNC | 463,766 |
| FLC | 20,569 |
| FLNC mean length | 2476 |
| FLNC/CCS | 77.01% |
| Items | Type | Results |
|---|---|---|
| Total nucleotides | Before correct | 98,097,845 |
| Total nucleotides | After correct | 98,043,063 |
| Total number | Before correct | 38,663 |
| Total number | After correct | 38,663 |
| Mean length | Before correct | 2538 |
| Mean length | After correct | 2536 |
| Min length | Before correct | 65 |
| Min length | After correct | 65 |
| Max length | Before correct | 11,283 |
| Max length | After correct | 11,283 |
| N50 | Before correct | 2699 |
| N50 | After correct | 2699 |
| N90 | Before correct | 1741 |
| N90 | After correct | 1741 |
| Transcripts Length Interval | <500 bp | 500–1 kbp | 1–2 kbp | 2–3 kbp | >3 kbp | Total |
|---|---|---|---|---|---|---|
| Number of transcripts | 324 | 1460 | 10,234 | 17,275 | 9370 | 38,663 |
| Number of genes | 183 | 891 | 5448 | 9445 | 6441 | 22,408 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.; Liao, X.; Han, T.; Chen, J.; He, C.; Lu, Z. Full-Length Transcriptomics Reveal the Gene Expression Profiles of Reef-Building Coral Pocillopora damicornis and Symbiont Zooxanthellae. Diversity 2021, 13, 543. https://doi.org/10.3390/d13110543
Guo Z, Liao X, Han T, Chen J, He C, Lu Z. Full-Length Transcriptomics Reveal the Gene Expression Profiles of Reef-Building Coral Pocillopora damicornis and Symbiont Zooxanthellae. Diversity. 2021; 13(11):543. https://doi.org/10.3390/d13110543
Chicago/Turabian StyleGuo, Zhuojun, Xin Liao, Tingyu Han, Junyuan Chen, Chunpeng He, and Zuhong Lu. 2021. "Full-Length Transcriptomics Reveal the Gene Expression Profiles of Reef-Building Coral Pocillopora damicornis and Symbiont Zooxanthellae" Diversity 13, no. 11: 543. https://doi.org/10.3390/d13110543
APA StyleGuo, Z., Liao, X., Han, T., Chen, J., He, C., & Lu, Z. (2021). Full-Length Transcriptomics Reveal the Gene Expression Profiles of Reef-Building Coral Pocillopora damicornis and Symbiont Zooxanthellae. Diversity, 13(11), 543. https://doi.org/10.3390/d13110543