
Homogenized Phylogeographic Structure across the Indo-Burma Ranges of a Large Monoecious Fig, Ficus altissima Blume


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Chloroplast DNA
2.3. nSSRs
2.4. Confirming Usability of nSSRs
2.5. Genetic Diversity
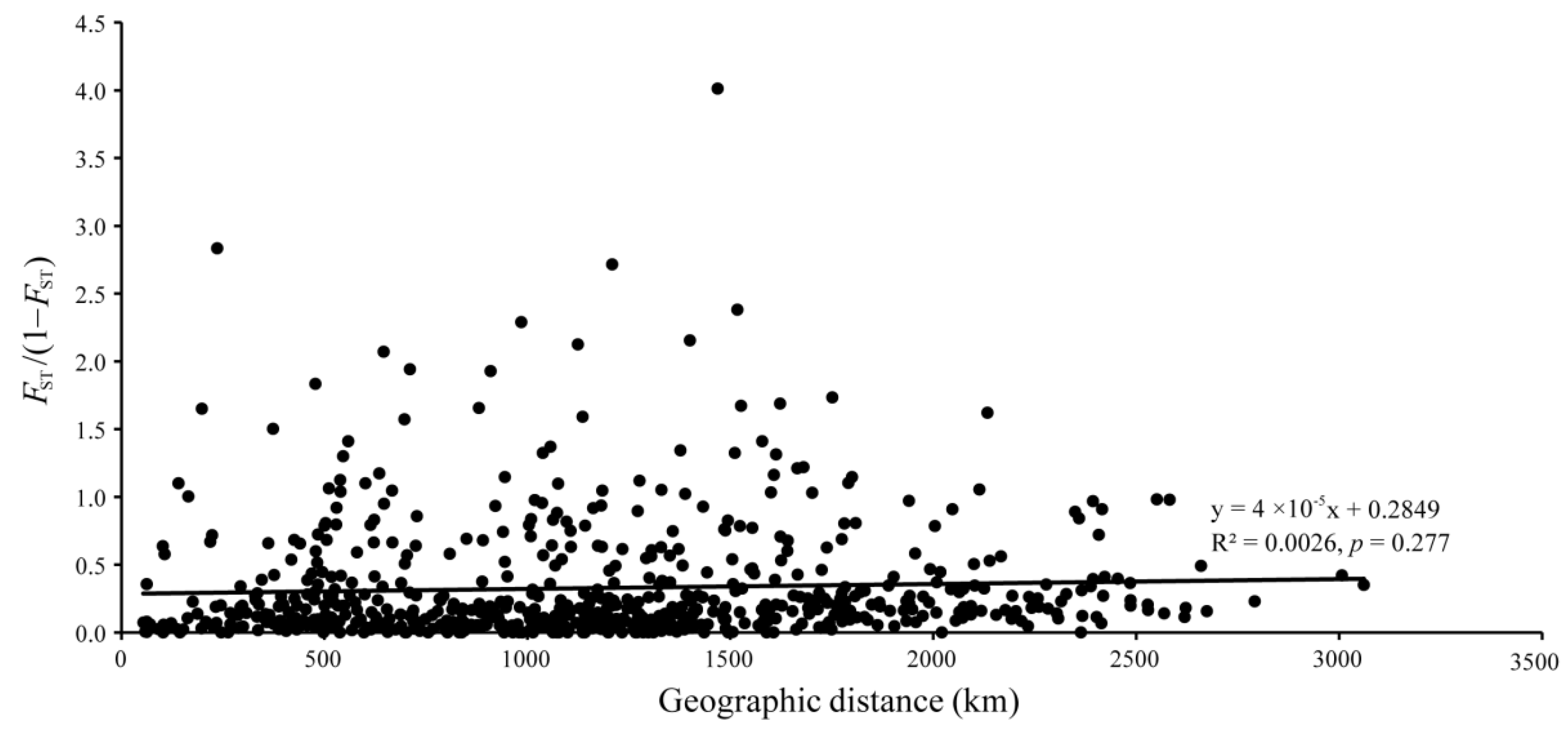
2.6. Genetic Structure
2.7. Population Dynamics
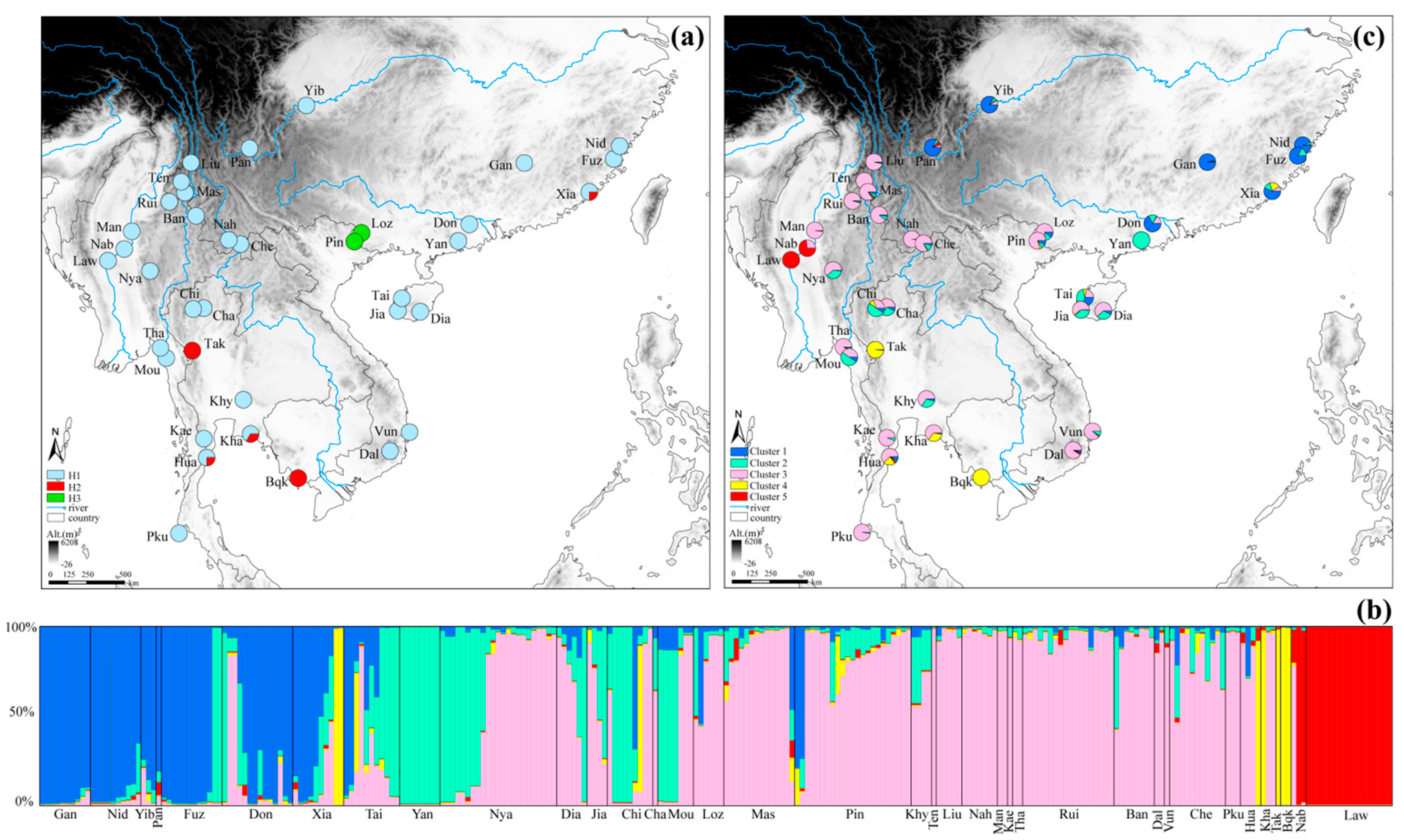
3. Results
3.1. cpDNA Genetic Diversity
3.2. Usability of nSSRs and Genetic Diversity
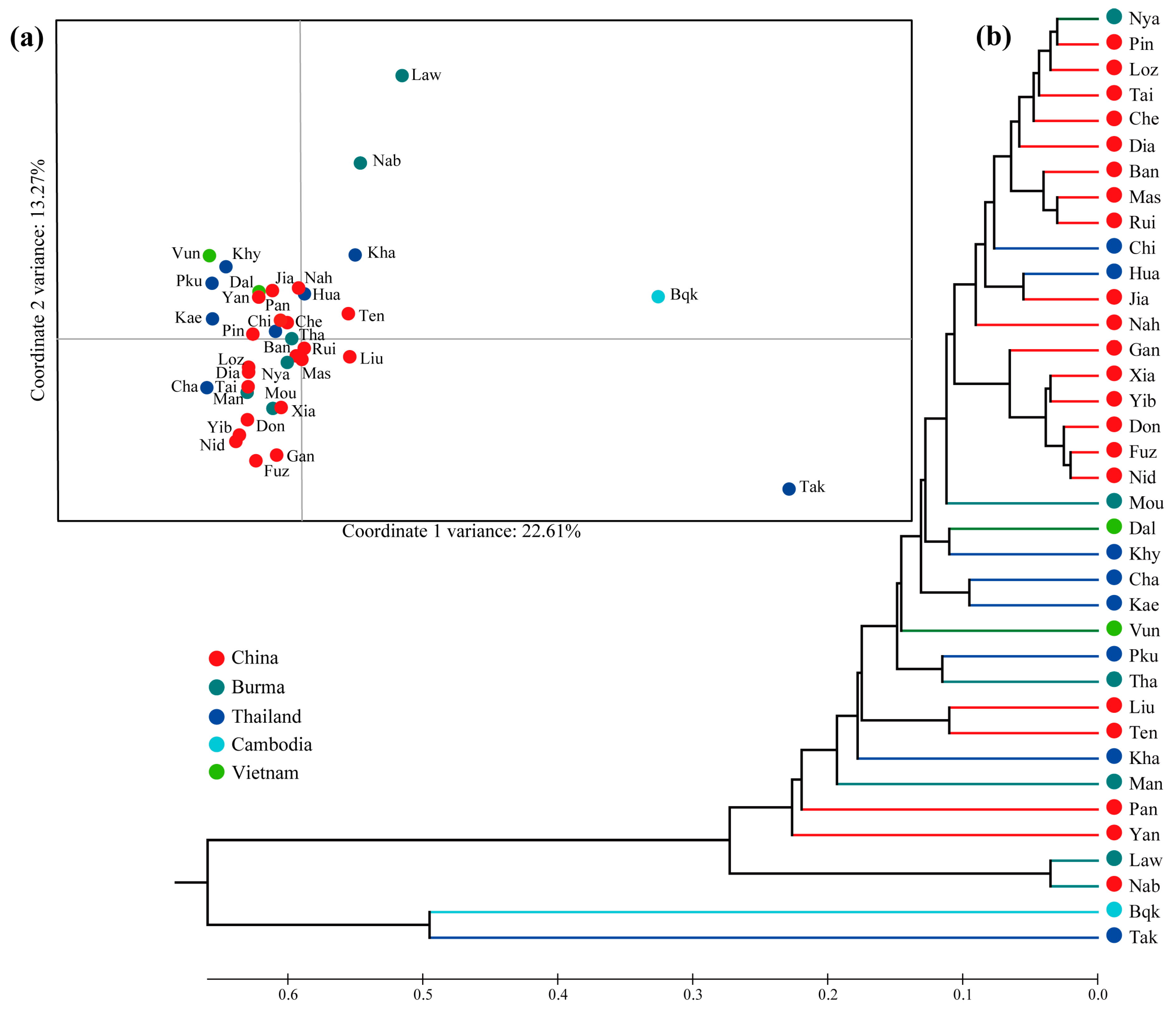
3.3. Population Structure
3.4. Population Dynamics
4. Discussion
4.1. Genetic Diversity, Population Dynamics, and Phylogeographic Patterns of Ficus altissima
4.2. Human Influence
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pop. | Country | Site | Lat. (N) | Long. (E) | cpDNA | nSSRs | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size | HD | π (10–2) | Size | Na | Np | PAr | HO | HE | FIS | |||||
| Yib | China | Yibin | 28.623 | 104.418 | 1 | na | na | 3 | 2.563 | 1 | 0.063 | 0.396 | 0.427 | 0.269 |
| Nid | Ningde | 26.661 | 119.532 | 6 | 0.000 | 0.000 | 10 | 2.938 | 0 | 0.000 | 0.556 | 0.428 | −0.252 | |
| Pan | Panzhihua | 26.554 | 101.680 | 1 | na | na | 1 | 1.438 | 0 | 0.000 | 0.438 | 0.219 | Na | |
| Fuz | Fuzhou | 26.153 | 119.291 | 7 | 0.000 | 0.000 | 12 | 2.750 | 0 | 0.000 | 0.469 | 0.435 | −0.033 | |
| Liu | Nujiang | 25.854 | 98.852 | 5 | 0.000 | 0.000 | 5 | 2.625 | 1 | 0.063 | 0.384 | 0.396 | 0.143 | |
| Gan | Ganzhou | 25.850 | 114.928 | 9 | 0.000 | 0.000 | 10 | 2.563 | 0 | 0.000 | 0.450 | 0.422 | −0.014 | |
| Ten | Tengchong | 24.943 | 98.387 | 1 | na | na | 1 | 1.375 | 0 | 0.000 | 0.375 | 0.188 | na | |
| Xia | Xiamen | 24.447 | 118.062 | 8 | 0.429 | 0.318 | 10 | 4.250 | 3 | 0.188 | 0.494 | 0.548 | 0.151 | |
| Mas | Mangshi | 24.414 | 98.566 | 14 | 0.000 | 0.000 | 14 | 4.125 | 0 | 0.000 | 0.470 | 0.478 | 0.056 | |
| Rui | Ruili | 23.976 | 97.810 | 18 | 0.000 | 0.000 | 18 | 4.000 | 1 | 0.063 | 0.465 | 0.518 | 0.130 | |
| Ban | Lincang | 23.297 | 99.099 | 8 | 0.000 | 0.000 | 8 | 3.500 | 0 | 0.000 | 0.485 | 0.516 | 0.127 | |
| Don | Donggang | 22.889 | 112.281 | 8 | 0.000 | 0.000 | 14 | 3.625 | 1 | 0.063 | 0.518 | 0.494 | −0.011 | |
| Loz | Chongzuo | 22.471 | 107.075 | 6 | 0.000 | 0.000 | 6 | 3.125 | 0 | 0.000 | 0.531 | 0.458 | −0.069 | |
| Nah | Jinghong | 22.131 | 100.675 | 7 | 0.000 | 0.000 | 7 | 3.188 | 2 | 0.125 | 0.509 | 0.474 | 0.008 | |
| Yan | Yangchun | 22.079 | 111.747 | 3 | 0.000 | 0.000 | 8 | 1.313 | 0 | 0.000 | 0.313 | 0.156 | −1.000 | |
| Pin | Pingxiang | 22.056 | 106.736 | 19 | 0.000 | 0.000 | 23 | 5.125 | 3 | 0.188 | 0.519 | 0.528 | 0.040 | |
| Che | Mengla | 21.926 | 101.240 | 11 | 0.000 | 0.000 | 11 | 3.688 | 0 | 0.000 | 0.494 | 0.509 | 0.076 | |
| Tai | Changjiang | 19.317 | 109.027 | 11 | 0.000 | 0.000 | 11 | 3.563 | 2 | 0.063 | 0.542 | 0.512 | −0.011 | |
| Jia | Ledong | 18.716 | 108.832 | 4 | 0.000 | 0.000 | 4 | 2.938 | 0 | 0.000 | 0.469 | 0.451 | 0.104 | |
| Dia | Baoting | 18.666 | 109.914 | 6 | 0.000 | 0.000 | 6 | 3.000 | 1 | 0.063 | 0.465 | 0.405 | −0.057 | |
| Man | Burma | Mandalay | 22.549 | 95.998 | 2 | 0.000 | 0.000 | 2 | 1.500 | 1 | 0.063 | 0.469 | 0.242 | −0.875 |
| Nab | Myotha | 21.697 | 95.635 | 3 | 0.000 | 0.000 | 3 | 2.125 | 0 | 0.000 | 0.479 | 0.396 | −0.011 | |
| Law | Bagan | 21.128 | 94.851 | 17 | 0.000 | 0.000 | 17 | 1.563 | 0 | 0.000 | 0.500 | 0.258 | −0.936 | |
| Nya | Taunggyi | 20.630 | 96.879 | 23 | 0.000 | 0.000 | 23 | 4.313 | 0 | 0.000 | 0.500 | 0.515 | 0.052 | |
| Tha | Thaton | 16.923 | 97.378 | 1 | na | na | 2 | 2.000 | 0 | 0.000 | 0.438 | 0.367 | 0.152 | |
| Mou | Moulmein | 16.433 | 97.658 | 7 | 0.000 | 0.000 | 7 | 2.563 | 0 | 0.000 | 0.469 | 0.369 | −0.197 | |
| Cha | Thailand | Lampang | 18.841 | 99.467 | 1 | na | na | 1 | 1.313 | 0 | 0.000 | 0.313 | 0.156 | na |
| Chi | Chiengmai | 18.795 | 98.962 | 5 | 0.000 | 0.000 | 9 | 3.813 | 2 | 0.125 | 0.500 | 0.495 | 0.048 | |
| Tak | Tak | 16.791 | 98.919 | 1 | na | na | 1 | 1.000 | 1 | 0.063 | 0.063 | 0.031 | na | |
| Khy | Nakhon Nayok | 14.413 | 101.375 | 4 | 0.000 | 0.000 | 4 | 2.000 | 0 | 0.000 | 0.563 | 0.352 | −0.500 | |
| Kha | Rayong | 12.766 | 101.728 | 3 | 0.667 | 0.495 | 3 | 3.250 | 2 | 0.125 | 0.563 | 0.580 | 0.229 | |
| Kae | Kaeng Krachan | 12.538 | 99.478 | 1 | na | na | 1 | 1.500 | 0 | 0.000 | 0.500 | 0.250 | na | |
| Hua | Thap Sakae | 11.625 | 99.615 | 4 | 0.500 | 0.371 | 4 | 3.313 | 3 | 0.188 | 0.365 | 0.540 | 0.451 | |
| Pku | Pkuket | 7.970 | 98.279 | 3 | 0.000 | 0.000 | 3 | 1.500 | 0 | 0.000 | 0.438 | 0.247 | −0.680 | |
| Vun | Vietnam | Phu Yen | 12.852 | 109.391 | 1 | na | na | 1 | 1.375 | 0 | 0.000 | 0.375 | 0.188 | na |
| Dal | Lam Dong | 11.940 | 108.458 | 2 | 0.000 | 0.000 | 2 | 2.375 | 0 | 0.000 | 0.531 | 0.438 | 0.128 | |
| Bqk | Cambodia | Bokor | 10.627 | 104.025 | 2 | 0.000 | 0.000 | 2 | 1.500 | 1 | 0.063 | 0.438 | 0.234 | −0.750 |
| Species | 233 | 0.245 | 0.104 | 267 | 8.500 | na | na | 0.483 | 0.576 | 0.163 | ||||
| Pop. | Nuclear Microsatellites | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fal_2 | Fal_9 | Fal_11 | Fal_14 | Fal_28 | Fal_31 | Fal_34 | Fal_41 | Fal_45 | Fal_46 | Fal_48 | Fal_50 | Fal_61 | Fal_62 | Fal_73 | Fal_75 | |
| Nid | no | No | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Fuz | no | no | 0.279 | no | no | no | no | 0.366 | no | no | no | no | no | no | no | no |
| Liu | no | no | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Gan | no | no | 0.304 | no | no | no | no | 0.340 | no | no | no | no | no | no | no | no |
| Xia | no | no | No | no | no | no | no | no | no | no | no | no | no | 0.342 | no | no |
| Mas | no | no | 0.298 | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Rui | no | 0.191 | 0.402 | no | no | no | no | no | no | no | no | no | 0.208 | no | no | no |
| Ban | no | no | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Don | no | no | 0.423 | no | no | no | no | 0.349 | no | no | no | no | no | no | no | no |
| Loz | no | no | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Nah | no | no | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Yan | no | no | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Pin | no | no | 0.117 | no | no | no | no | 0.228 | no | no | no | no | 0.143 | no | no | no |
| Che | no | no | 0.379 | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Tai | no | no | 0.186 | no | no | no | no | 0.375 | no | no | no | no | no | no | no | no |
| Jia | no | no | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Dia | no | no | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Law | no | no | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Nya | no | no | 0.272 | no | no | no | no | no | no | no | no | no | 0.250 | no | no | no |
| Mou | no | no | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
| Chi | no | 0.243 | No | no | no | no | no | 0.296 | no | no | no | no | no | no | no | no |
| Khy | no | no | No | no | no | no | no | no | no | no | no | no | no | no | no | no |
References
- Mittermeier, R.A.; Gil, P.R.; Hoffman, M.; Pilgrim, J.; Brooks, T.; Mittermeier, C.G.; Lamoreux, J.; de Fonseca, G.A.B. Hotspots Revisited: Earth’s Biologically Richest and Most Endangered Terrestrial Ecoregions; CEMEX: Mexico City, Mexico, 2004. [Google Scholar]
- Meng, H.H.; Gao, X.Y.; Song, Y.G.; Cao, G.L.; Li, J. Biodiversity arks in the Anthropocene. Reg. Sustain. 2021, 2, 109–115. [Google Scholar] [CrossRef]
- Voris, H.K. Maps of pleistocene sea levels in Southeast Asia: Shorelines, river systems, time durations. J. Biogeogr. 2000, 27, 1153–1167. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, D.S. Biogeography and conservation in Southeast Asia: How 2.7 million years of repeated environmental fluctuations affect today’s patterns and the future of the remaining refugial-phase biodiversity. Biodivers. Conserv. 2010, 19, 919–941. [Google Scholar] [CrossRef] [Green Version]
- Lukoschek, V.; Osterhage, J.L.; Karns, D.R.; Murphy, J.C.; Voris, H.K. Phylogeography of the Mekong mud snake (Enhydris subtaeniata): The biogeographic importance of dynamic river drainages and fluctuating sea levels for semiaquatic taxa in Indochina. Ecol. Evol. 2011, 1, 330–342. [Google Scholar] [CrossRef]
- Sodhi, N.S.; Koh, L.P.; Brook, B.W.; Ng, P.K. Southeast Asian biodiversity: An impending disaster. Trends Ecol. Evol. 2004, 19, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Nijiman, V. An overview of international wildlife trade from Southeast Asia. Biodivers. Conserv. 2010, 19, 1101–1114. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.C. Have Indo-Malaysian forests reached the end of the road? Biol. Conserv. 2018, 223, 129–137. [Google Scholar] [CrossRef]
- Fuchs, J.; Ericson, P.G.; Pasquet, E. Mitochondrial phylogeographic structure of the white-browed piculet (Sasia ochracea): Cryptic genetic differentiation and endemism in Indochina. J. Biogeogr. 2008, 35, 565–575. [Google Scholar] [CrossRef]
- Hartvig, I.; So, T.; Changtragoon, S.; Tran, H.T.; Bouamanivong, S.; Theilade, I.; Kjaer, E.D.; Nielsen, L.R. Population genetic structure of the endemic rosewoods Dalbergia cochinchinensis and D. oliveri at a regional scale reflects the Indochinese landscape and life-history traits. Ecol. Evol. 2018, 8, 530–545. [Google Scholar] [CrossRef] [Green Version]
- Attwood, S.W. Schistosomiasis in the Mekong region: Epidemiology and phylogeography. Adv. Parasitol. 2001, 50, 87–152. [Google Scholar]
- Thaenkham, U.; Phuphisut, O.; Nuamtanong, S.; Yoonuan, T.; Sa-Nguankiat, S.; Vonghachack, Y.; Belizario, V.Y.; Dung, D.T.; Dekumyoy, P.; Waikagul, J. Genetic differences among Haplorchis taichui populations in Indochina revealed by mitochondrial COX1 sequences. J. Helminthol. 2017, 91, 597–604. [Google Scholar] [CrossRef]
- Pramual, P.; Kuvangkadilok, C.; Baimai, V.; Walton, C. Phylogeography of the black fly Simulium tani (Diptera: Simuliidae) from Thailand as inferred from mtDNA sequences. Mol. Ecol. 2005, 14, 3989–4001. [Google Scholar] [CrossRef] [PubMed]
- Takagi, A.P.; Ishikawa, S.; Nao, T.; Song, S.L.; Hort, S.; Thammavong, K.; Saphakdy, B.; Phomsouvanhm, A.; Nishida, M.; Kurokura, H. Genetic differentiation and distribution routes of the bronze featherback Notopterus notopterus (Osteoglossiformes: Notopteridae) in Indochina. Biol. J. Linnean Soc. 2010, 101, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Adamson, E.A.; Hurwood, D.A.; Mather, P.B. Insights into historical drainage evolution based on the phylogeography of the chevron snakehead fish (Channa striata) in the Mekong Basin. Freshw. Biol. 2012, 57, 2211–2229. [Google Scholar] [CrossRef]
- Blair, C.; Davy, C.M.; Ngo, A.; Orlov, N.L.; Shi, H.T.; Lu, S.Q.; Gao, L.; Rao, D.Q.; Murphy, R.W. Genealogy and demographic history of a widespread amphibian throughout Indochina. J. Hered. 2013, 104, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meschersky, I.G.; Abramov, A.V.; Lebedev, V.S.; Chichkina, A.N.; Rozhnov, V.V. Evidence of a complex phylogeographic structure in the Indomalayan pencil-tailed tree mouse Chiropodomys gliroides (Rodentia: Muridae) in eastern Indochina. Biocheml. Syst. Ecol. 2016, 65, 147–157. [Google Scholar] [CrossRef]
- Balakirev, A.E.; Rozhnov, V.V. Taxonomic revision of beautiful squirrels (Callosciurus, Rodentia: Sciuridae) from the Callosciurus erythraeus/finlaysonii complex and their distribution in eastern Indochina. Raffles Bull. Zool. 2019, 67, 459–489. [Google Scholar]
- Balakrishnan, C.N.; Monfort, S.L.; Gaur, A.; Singh, L.; Sorenson, M.D. Phylogeography and conservation genetics of Eld’s deer (Cervus eldi). Mol. Ecol. 2003, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Glaubrecht, M.; Köhler, F. Radiating in a river: Systematics, molecular genetics and morphological differentiation of viviparous freshwater gastropods endemic to the Kaek River, central Thailand (Cerithioidea, Pachychilidae). Biol. J. Linnean Soc. 2004, 82, 275–311. [Google Scholar] [CrossRef] [Green Version]
- McGill, B.J.; Dornelas, M.; Gotelli, N.J.; Magurran, A.E. Fifteen forms of biodiversity trend in the Anthropocene. Trends Ecol. Evol. 2015, 30, 104–113. [Google Scholar] [CrossRef]
- Newbold, T.; Hudson, L.N.; Hill, S.L.L.; Contu, S.; Lysenko, I.; Senior, R.A.; Börger, L.; Bennett, D.J.; Choimes, A.; Collen, B.; et al. Global effects of land use on local terrestrial biodiversity. Nature 2015, 520, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Tilman, D.; Clark, M.; Williams, D.R.; Kimmel, K.; Polasky, S.; Packer, C. Future threats to biodiversity and pathways to their prevention. Nature 2017, 546, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Herre, E.A.; Jander, K.C.; Machado, C.A. Evolutionary ecology of figs and their associates: Recent progress and outstanding puzzles. Annu. Rev. Ecol. Evol. Syst. 2008, 39, 439–458. [Google Scholar] [CrossRef] [Green Version]
- Cruaud, A.; Rønsted, N.; Chantarasuwan, B.; Chou, L.S.; Clement, W.L.; Couloux, A.; Cousins, B.; Genson, G.; Harrison, R.D.; Hanson, P.E.; et al. An extreme case of plant–insect co-diversification: Figs and fig-pollinating wasps. Syst. Biol. 2012, 61, 1029–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Mon, A.M.; Fu, Y.; Zhang, Y.; Wang, C.; Yang, X.; Wang, Y. The genus Ficus (Moraceae) used in diet: Its plant diversity, distribution, traditional uses and ethnopharmacological importance. J. Ethnopharmacol. 2018, 226, 185–196. [Google Scholar] [CrossRef]
- Peng, Y.Q.; Duan, Z.B.; Yang, D.R.; Rasplus, J.Y. Co-occurrence of two Eupristina species on Ficus altissima in Xishuangbanna, SW China. Symbiosis 2008, 45, 9–14. [Google Scholar]
- Khare, C.P. Indian Medicinal Plants: An Illustrated Dictionary; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Berg, C.C.; Corner, E.G.H. Moraceae–Ficus. In Flora Malesiana; Nooteboom, H.P., Ed.; National Herbarium Nederland: Leiden, The Netherlands, 2005. [Google Scholar]
- Peng, Y.Q.; Compton, S.G.; Yang, D.R. The reproductive success of Ficus altissima and its pollinator in a strongly seasonal environment: Xishuangbanna, Southwestern China. Plant Ecol. 2010, 209, 227–236. [Google Scholar] [CrossRef]
- Zhou, Z.K.; Gilbert, M.G. Moraceae: Genus Ficus. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St Louis, MO, USA, 2003; Volume 5, pp. 37–71. [Google Scholar]
- Fang, J.Y.; Wang, Z.H.; Tang, Z.Y. Atlas of Woody Plants in China: Distribution and Climate; Higher Education Press: Beijing, China; Springer-Verlag: Berlin, Germany, 2011; Volume 1, p. 203. [Google Scholar]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, F.D.A.; Santana, J.D.S.; Dos Santos, R.M.; Fajardo, C.G.; Coelho, G.D.O.; Carvalho, D.D. DNA extraction protocols and cpDNA primers to Ficus bonijesulapensis (Moraceae). Caatinga 2010, 23, 69–74. [Google Scholar]
- Honorio Coronado, E.N.; Dexter, K.G.; Poelchau, M.F.; Hollingsworth, P.M.; Phillips, O.L.; Pennington, R.T. Ficus insipida subsp. insipida (Moraceae) reveals the role of ecology in the phylogeography of widespread Neotropical rain forest tree species. J. Biogeogr. 2014, 41, 1697–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tnah, L.H.; Lee, S.L.; Tan, A.L.; Lee, C.T.; Ng, K.K.S.; Ng, C.H. Intraspecific classification of Ficus deltoidea Jack subsp. deltoidea (Moraceae) in Peninsular Malaysia based on morphological and molecular variations. Biochem. Syst. Ecol. 2016, 67, 119–128. [Google Scholar] [CrossRef]
- Costa, P.C.; Lorenz-Lemke, A.P.; Furini, P.R.; Honorio Coronado, E.N.; Kjellberg, F.; Pereira, R.A. The phylogeography of two disjunct Neotropical Ficus (Moraceae) species reveals contrasted histories between the Amazon and the Atlantic Forests. Bot. J. Linnean Soc. 2017, 185, 272–289. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Emshwiller, E.; Doyle, J.J. Chloroplast-expressed glutamine synthetase (ncpGS): Potential utility for phylogenetic studies with an example from Oxalis (Oxalidaceae). Mol. Phylogenet. Evol. 1999, 12, 310–319. [Google Scholar] [CrossRef]
- Rønsted, N.; Weiblen, G.; Clement, W.L.; Zerega, N.J.C.; Savolainen, V. Reconstructing the phylogeny of figs (Ficus, Moraceae) to reveal the history of the fig pollination mutualism. Symbiosis 2008, 45, 45–55. [Google Scholar]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 1999, 6, 365–386. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Rousset, F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.; Smouse, P.E. GenAIEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goudet, J. FSTAT, A Program to Estimate and Test Gene Diversities and Fixation Indices, Version 2.9.3; ScienceOpen: Burlington, MA, USA, 2001. [Google Scholar]
- Marshall, T.C.; Slate, J.; Kruuk, L.E.B.; Pemberton, J.M. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 1998, 7, 639–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Hengl, T.A. Practical Guide to Geostatistical Mapping; University of Amsterdam: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Ersts, P.J. Geographic Distance Matrix Generator (Version 1.2.3). Available online: http://biodiversityinformatics.amnh.org/open_source/gdmg (accessed on 21 July 2021).
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Piry, S.G.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting reductions in the effective population size using allele frequency data. J. Hered. 1999, 86, 502–503. [Google Scholar] [CrossRef]
- Di Rienzo, A.; Peterson, A.C.; Garza, J.C.; Valdes, A.M.; Slatkin, M.; Freimer, N.B. Mutational processes of simple sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 1994, 91, 3166–3170. [Google Scholar] [CrossRef] [Green Version]
- Luikart, G.; Cornuet, J.M. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv. Biol. 1998, 12, 228–237. [Google Scholar] [CrossRef]
- Nei, M.; Maruyama, T.; Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef]
- Luikart, G.L.; Allendorf, F.W.; Cornuet, J.M.; Sherwin, W.B. Distortion of allele frequency distributions provides a test for recent populations. J. Hered. 1998, 89, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Vieira, F.D.A.; Novaes, R.M.L.; Fajardo, C.G.; Santos, R.M.D.; Almeida, H.D.S.; Carvalho, D.D.; Lovato, M.B. Holocene southward expansion in seasonally dry tropical forests in South America: Phylogeography of Ficus bonijesulapensis (Moraceae). Bot. J. Linnean Soc. 2015, 177, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Q.; Chen, J.Y.; Wang, S.; Xiong, S.Z. Evaluation of six candidate DNA barcoding loci in Ficus (Moraceae) of China. Mol. Ecol. Resour. 2012, 12, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A. The shapes of neutral gene genealogies in two species: Probabilities of monophyly, paraphyly, and polyphyly in a coalescent model. Evolution 2003, 57, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Lomáscolo, S.B.; Levey, D.J.; Kimball, R.T.; Bolker, B.M.; Alborn, H.T. Dispersers shape fruit diversity in Ficus (Moraceae). Proc. Natl. Acad. Sci. USA 2010, 107, 14668–14672. [Google Scholar] [CrossRef] [Green Version]
- Harrison, R.D.; Hamid, A.A.; Kenta, T.; Lafrankie, J.; Lee, H.S.; Nagamasu, H.; Nakashizuka, T.; Palmiotto, P. The diversity of hemi-epiphytic figs (Ficus; Moraceae) in a Bornean lowland rain forest. Biol. J. Linnean Soc. 2003, 78, 439–455. [Google Scholar] [CrossRef] [Green Version]
- Compton, S.G.; Ellwood, M.D.; Davis, A.J.; Welch, K. The flight heights of chalcid wasps (Hymenoptera, Chalcidoidea) in a lowland Bornean rain forest: Fig wasps are the high fliers. Biotropica 2000, 32, 515–522. [Google Scholar] [CrossRef]
- Harrison, R.D. Fig wasp dispersal and the stability of a keystone plant resource in Borneo. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, S76–S79. [Google Scholar] [CrossRef] [Green Version]
- Bain, A.; Borges, R.M.; Chevallier, M.H.; Vignes, H.; Kobmoo, N.; Peng, Y.Q.; Cruaud, A.; Rasplus, J.Y.; Kjellberg, F.; Hossaert-Mckey, M. Geographic structuring into vicariant species-pairs in a wide-ranging, high-dispersal plant–insect mutualism: The case of Ficus racemosa and its pollinating wasps. Evol. Ecol. 2016, 30, 663–684. [Google Scholar] [CrossRef]
- Nason, J.D.; Herre, E.A.; Hamrick, J.L. The breeding structure of a tropical keystone plant resource. Nature 1998, 391, 685–687. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Compton, S.G.; Butlin, R.K.; Gilmartin, P.M. Wind-borne insects mediate directional pollen transfer between desert fig trees 160 kilometers apart. Proc. Natl. Acad. Sci. USA 2009, 106, 20342–20347. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.Y.; Chou, L.S.; Di Giusto, B.; Bain, A. Sexual specialization in phenology in dioecious Ficus benguetensis and its consequences for the mutualism. Bot. Stud. 2015, 56, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, N.; McKey, D.; Kjellberg, F.; Hossaert-McKey, M. Phylogeography and historical biogeography of obligate specific mutualisms. In The Biogeography of Host–Parasite Interactions; Morand, S., Krasnov, B.R., Eds.; Oxford University Press: Oxford, UK, 2010; pp. 31–39. [Google Scholar]
- Yu, H.; Nason, J.D. Nuclear and chloroplast DNA phylogeography of Ficus hirta: Obligate pollination mutualism and constraints on range expansion in response to climate change. New Phytol. 2013, 197, 276–289. [Google Scholar] [CrossRef]
- Kjellberg, F.; Valdeyron, G. Species-specific pollination: A help or a limitation to range extension. In Biological Invasions in Europe and the Mediterranean Basin; di Castri, F., Hansen, A.J., Eds.; Dr W Junk publishers: Dordrecht, The Netherlands, 1990; pp. 371–378. [Google Scholar]
- Xu, L.; Harrison, R.D.; Yang, P.; Yang, D.R. New insight into the phylogenetic and biogeographic history of genus Ficus: Vicariance played a relatively minor role compared with ecological opportunity and dispersal. J. Syst. Evol. 2011, 49, 546–557. [Google Scholar] [CrossRef]
- Muller, J. Palynological evidence for change in geomorphology, climate and vegetation in the Mio-Pliocene of Malesia. In Transactions of the Second Aberdeen-Hull Symposium on Malesian Ecology; Ashton, P., Ashton, M., Eds.; University of Hull: Hull, UK, 1972; pp. 6–16. [Google Scholar]
- Morley, R.J. Origin and Evolution of Tropical Rain Forests; John Wiley and Sons Ltd.: Chichester, UK, 2000. [Google Scholar]
- Heaney, L.R. A synopsis of climatic and vegetational change in Southeast Asia. Clim. Chang. 1991, 19, 53–61. [Google Scholar] [CrossRef]
- Tordoff, A.W.; Bezuijen, M.R.; Duckworth, J.W.; Fellowes, J.R.; Koenig, K.; Pollard, E.H.B.; Royo, A.G. Ecosystem Profile: Indo-Burma Biodiversity Hotspot 2011 Update; Critical Ecosystem Partnership Fund: Arlington, VA, USA, 2012. [Google Scholar]
- Hubbell, S.P.; Foster, R.B. Diversity of canopy trees in a neotropical forest and implications for conservation. In Tropical Rain Forest: Ecology and Management; Sutton, S.L., Whitmore, T.C., Chadwick, A.C., Eds.; Blackwell Scientific Publications: Oxford, UK, 1983. [Google Scholar]
- Fedorov, A.A. The structure of the tropical rain forest and speciation in the humid tropics. J. Ecol. 1966, 54, 1–11. [Google Scholar] [CrossRef]
- Dick, C.W.; Hardy, O.J.; Jones, F.A.; Petit, R.J. Spatial scales of pollen and seed-mediated gene flow in tropical rain forest trees. Trop. Plant Biol. 2008, 1, 20–33. [Google Scholar] [CrossRef]
- Dick, C.W.; Heuertz, M. The complex biogeographic history of a widespread tropical tree species. Evolution 2008, 62, 2760–2774. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhang, J.; Chen, Y.; Compton, S.G.; Chen, X. Contrasting genetic responses to population fragmentation in a coevolving fig and fig wasp across a mainland-island archipelago. Mol. Ecol. 2013, 22, 4384–4396. [Google Scholar] [CrossRef] [PubMed]
- Colabella, F.; Gallo, L.A.; Moreno, A.C.; Marchelli, P. Extensive pollen flow in a natural fragmented population of Patagonian cypress Austrocedrus chilensis. Tree Genet. Genomes 2014, 10, 1519–1529. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, Z.X.; Compton, S.G.; Liu, M.; Chen, X.Y. Genetic diversity and differentiation of the extremely dwarf Ficus tikoua in Southwestern China. Biochem. Syst. Ecol. 2011, 39, 441–448. [Google Scholar] [CrossRef]
- Liu, M.; Compton, S.G.; Peng, F.E.; Zhang, J.; Chen, X.Y. Movements of genes between populations: Are pollinators more effective at transferring their own or plant genetic markers? Proc. R. Soc. Lond. B Biol. Sci. 2015, 282, 20150290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, E.; Nason, J.D.; Machado, C.A.; Zheng, L.; Yu, H.; Kjellberg, F. Lack of genetic isolation by distance, similar genetic structuring but different demographic histories in a fig-pollinating wasp mutualism. Mol. Ecol. 2015, 24, 5976–5991. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.Y.; Van Noort, S.; Compton, S.G.; Chen, Y.; Greeff, J.M. The genetic consequences of habitat specificity for fig trees in southern African fragmented forests. Acta Oecol. 2020, 102, 103506. [Google Scholar] [CrossRef]
- Corlett, R.T. The Anthropocene concept in ecology and conservation. Trends Ecol. Evol. 2015, 30, 36–41. [Google Scholar] [CrossRef]
- Crutzen, P.J.; Stoermer, E.F. The ‘Anthropocene’. IGBP Newsl. 2000, 41, 17–18. [Google Scholar]
- Isbel, F.; Gonzalez, A.; Loreau, M.; Cowles, J.; Díaz, S.; Hector, A.; Mace, G.M.; Wardle, D.A.; O’Connor, M.I.; Duffy, J.E.; et al. Linking the influence and dependence of people on biodiversity across scales. Nature 2017, 546, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.N.; Balmford, A.; Brook, B.W.; Buettel, J.C.; Galetti, M.; Guangchun, L.; Wilmshurst, J.A. Biodiversity losses and conservation responses in the Anthropocene. Science 2017, 356, 270–275. [Google Scholar] [CrossRef]
- Chen, C.; Li, P.; Wang, R.H.; Schaal, B.A.; Fu, C.X. The population genetics of cultivation: Domestication of a traditional Chinese medicine, Scrophularia ningpoensis Hemsl. (Scrophulariaceae). PLoS ONE 2014, 9, e105064. [Google Scholar]
- Eyre-Walker, A.; Gaut, R.L.; Hilton, H.; Feldman, D.L.; Gaut, B.S. Investigation of the bottleneck leading to the domestication of maize. Proc. Natl. Acad. Sci. USA 1998, 95, 4441–4446. [Google Scholar] [CrossRef] [Green Version]
- Londo, J.P.; Chiang, Y.C.; Hung, K.H.; Chiang, T.Y.; Schaal, B.A. Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza Sativa. Proc. Natl. Acad. Sci. USA 2006, 103, 9578–9583. [Google Scholar] [CrossRef] [Green Version]
- Sonnante, G.; Stockton, T.; Nodari, R.O.; Becerra Velásquez, V.L.; Gepts, P. Evolution of genetic diversity during the domestication of common-bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 1994, 89, 629–635. [Google Scholar] [CrossRef]
- Hu, H.B. Sacred natural sites in Xishuangbanna, south-western China. In The Importance of Sacred Natural Sites for Biodiversity Conservation; Lee, C., Schaaf, T., Eds.; United Nations Educational, Scientific and Cultural Organization: Paris, France, 2003; pp. 127–133. [Google Scholar]




| Locus | Primer Sequences | Repeat Motif | GenBank Accession No. | PIC |
|---|---|---|---|---|
| Fal_2 | F: CCTGTGGGAGAGTTTGAAGG | (CGT)6 | MN255360 | 0.487 |
| R: CTTGCTGCACGAATCTGCT | ||||
| Fal_9 | F: GAGTACATGCAAATGCCTCG | (TTTA)5 | MN255361 | 0.561 |
| R: CTCAGCAGCAACGAAAGATG | ||||
| Fal_11 | F: GACCTGTTGGAGGAGATTGC | (GCG)5 | MN255362 | 0.753 |
| R: TCATGGGCCACTTATCCTTC | ||||
| Fal_14 | F: CGATCCTTATCCTCTGCTCG | (AAG)10 | MN255363 | 0.740 |
| R: GCACGCAATTTGAACGAAC | ||||
| Fal_28 | F: TCAGAATTGGAACGAGGGAC | (TTTG)7 | MN255364 | 0.492 |
| R: GCAGGGACTTCTTCTCTGACC | ||||
| Fal_31 | F: CGATCACCACGAGCTACTGA | (GTCT)5 | MN255365 | 0.803 |
| R: TGGCGCATGATAAGTTTGAG | ||||
| Fal_34 | F: CCAACTAGCCACACTTTGGA | (AATCCC)5 | MN255366 | 0.582 |
| R: TGGCACAATTGACCTCAGAA | ||||
| Fal_41 | F: CTCTTGGATACCGAGTCCGA | (TTTG)5 | MN255367 | 0.434 |
| R: GGACTGAACTGCTGTCATGTG | ||||
| Fal_45 | F: TCGAAATCGGATACTCCTCG | (TTTTAT)5 | MN255368 | 0.583 |
| R: CATGAAGCTTGAGCATTGGA | ||||
| Fal_46 | F: GCCACGACATCACATCATTA | (ACAT)6 | MN255369 | 0.444 |
| R: TCAGCTTACCTTATTGGCCG | ||||
| Fal_48 | F: ATGTGCCAAACCCAGAACTC | (AAAC)5 | MN255370 | 0.764 |
| R: CAACCTAGCTCTCGGAGGTG | ||||
| Fal_50 | F: GCCCATCTGGTGACTGAAAC | (AAT)7 | MN255371 | 0.351 |
| R: CGTGTGCATGCTTCATCTCT | ||||
| Fal_61 | F: TGGGCTCGTGACTGACTAGA | (TTG)6 | MN255372 | 0.137 |
| R: ATGTGGGGACGGCCTCTT | ||||
| Fal_62 | F: CACGTGGTGGCTATGTTCTG | (TTA)7 | MN255373 | 0.215 |
| R: GCTACGGTTTATTTGCGGTG | ||||
| Fal_73 | F: ATCCTTTGCTTTGCTCGTGT | (ATA)9 | MN255374 | 0.813 |
| R: CGAACCTTGCACACCCTAAT | ||||
| Fal_75 | F: GGATCCAAAATTGGGCAGT | (AAT)8 | MN255375 | 0.459 |
| R: ATTCATGGAATCATGGGCAC |
| Source of Variation | df | SS | VC | PV (%) | FST | |
|---|---|---|---|---|---|---|
| cpDNA | Among populations | 36 | 305.621 | 1.37 | 95.83 | |
| Within populations | 196 | 11.667 | 0.06 | 4.17 | ||
| Total | 232 | 317.288 | 1.43 | FST = 0.958 | ||
| nSSRs | Among populations | 36 | 554.386 | 0.82 | 17.75 | |
| Within populations | 497 | 1888.006 | 3.80 | 82.25% | ||
| Total | 533 | 2442.391 | 4.62 | FST = 0.178 |
| Population | Size | Model Shift | TPM | SMM |
|---|---|---|---|---|
| p Value | p Value | |||
| Che | 11 | normal L-shaped distribution | 0.337 | 0.448 |
| Don | 14 | normal L-shaped distribution | 0.047 * | 0.336 |
| Fuz | 12 | normal L-shaped distribution | 0.033 * | 0.245 |
| Gan | 10 | shifted mode | 0.010 * | 0.073 |
| Law | 17 | shifted mode | 0.002 ** | 0.005 ** |
| Mas | 14 | normal L-shaped distribution | 0.031 * | 0.0002 |
| Nid | 10 | shifted mode | 0.233 | 0.503 |
| Nya | 23 | normal L-shaped distribution | 0.366 | 0.008 ** |
| Pin | 23 | normal L-shaped distribution | 0.540 | 0.024 * |
| Rui | 18 | normal L-shaped distribution | 0.355 | 0.078 |
| Tai | 11 | normal L-shaped distribution | 0.133 | 0.566 |
| Xia | 10 | shifted mode | 0.551 | 0.075 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.-F.; Darwell, C.T.; Peng, Y.-Q. Homogenized Phylogeographic Structure across the Indo-Burma Ranges of a Large Monoecious Fig, Ficus altissima Blume. Diversity 2021, 13, 654. https://doi.org/10.3390/d13120654
Huang J-F, Darwell CT, Peng Y-Q. Homogenized Phylogeographic Structure across the Indo-Burma Ranges of a Large Monoecious Fig, Ficus altissima Blume. Diversity. 2021; 13(12):654. https://doi.org/10.3390/d13120654
Chicago/Turabian StyleHuang, Jian-Feng, Clive T. Darwell, and Yan-Qiong Peng. 2021. "Homogenized Phylogeographic Structure across the Indo-Burma Ranges of a Large Monoecious Fig, Ficus altissima Blume" Diversity 13, no. 12: 654. https://doi.org/10.3390/d13120654
APA StyleHuang, J. -F., Darwell, C. T., & Peng, Y. -Q. (2021). Homogenized Phylogeographic Structure across the Indo-Burma Ranges of a Large Monoecious Fig, Ficus altissima Blume. Diversity, 13(12), 654. https://doi.org/10.3390/d13120654