
Chromosomal-Level Assembly of Antarctic Scaly Rockcod, Trematomus loennbergii Genome Using Long-Read Sequencing and Chromosome Conformation Capture (Hi-C) Technologies

 , ,
, ,  , , ,
, , , 
Abstract
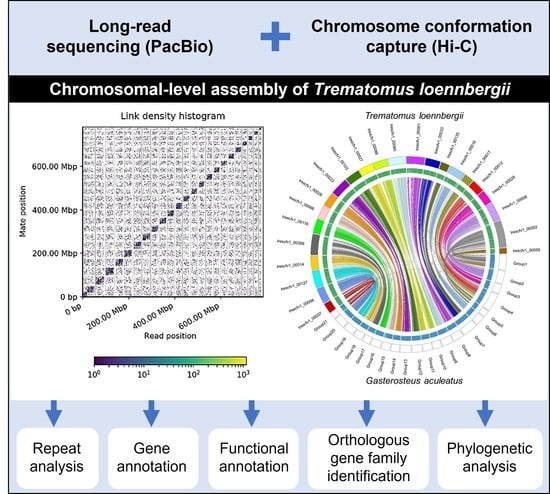
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Genome Sequencing and Assembly Using PacBio Long Reads
2.3. Hi-C Analysis and Chromosome Assembly
2.4. Quality Evaluation
2.5. Transcriptome Sequencing
2.6. Genome Annotation and Repeat Analysis
2.7. Functional Annotation
2.8. Gene Family Identification and Phylogenetic Analysis
3. Results and Discussion
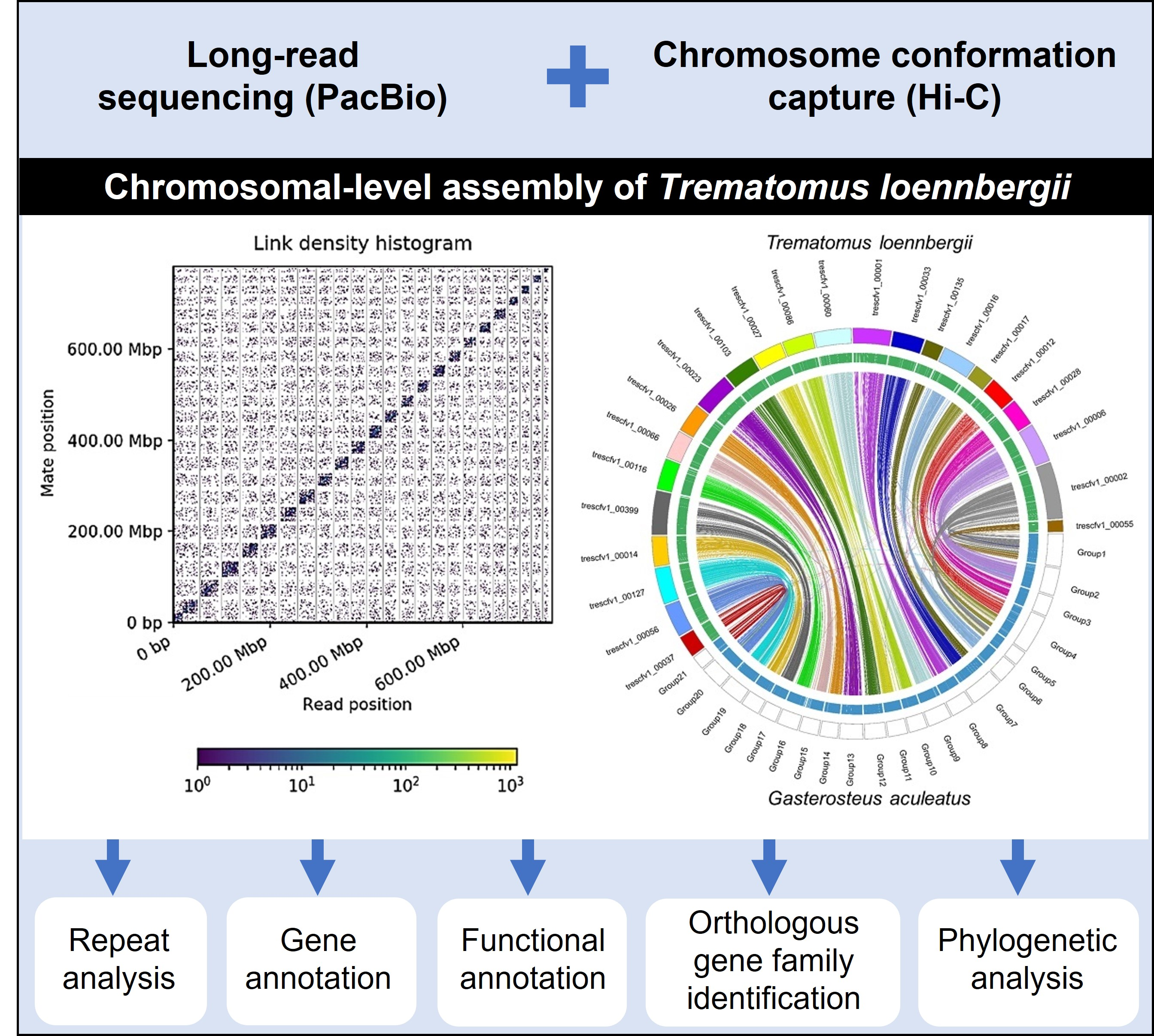
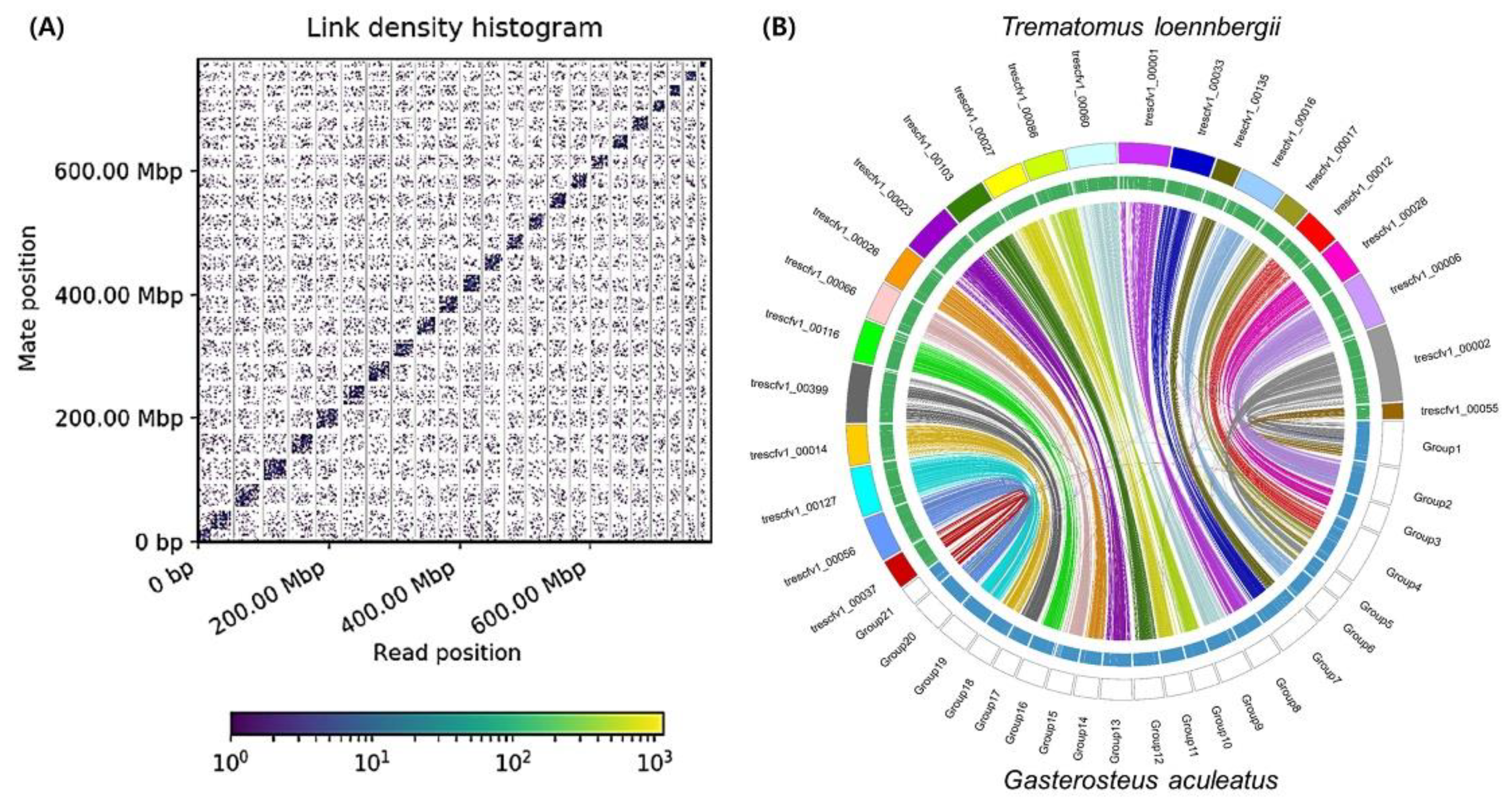
3.1. Genome Assembly
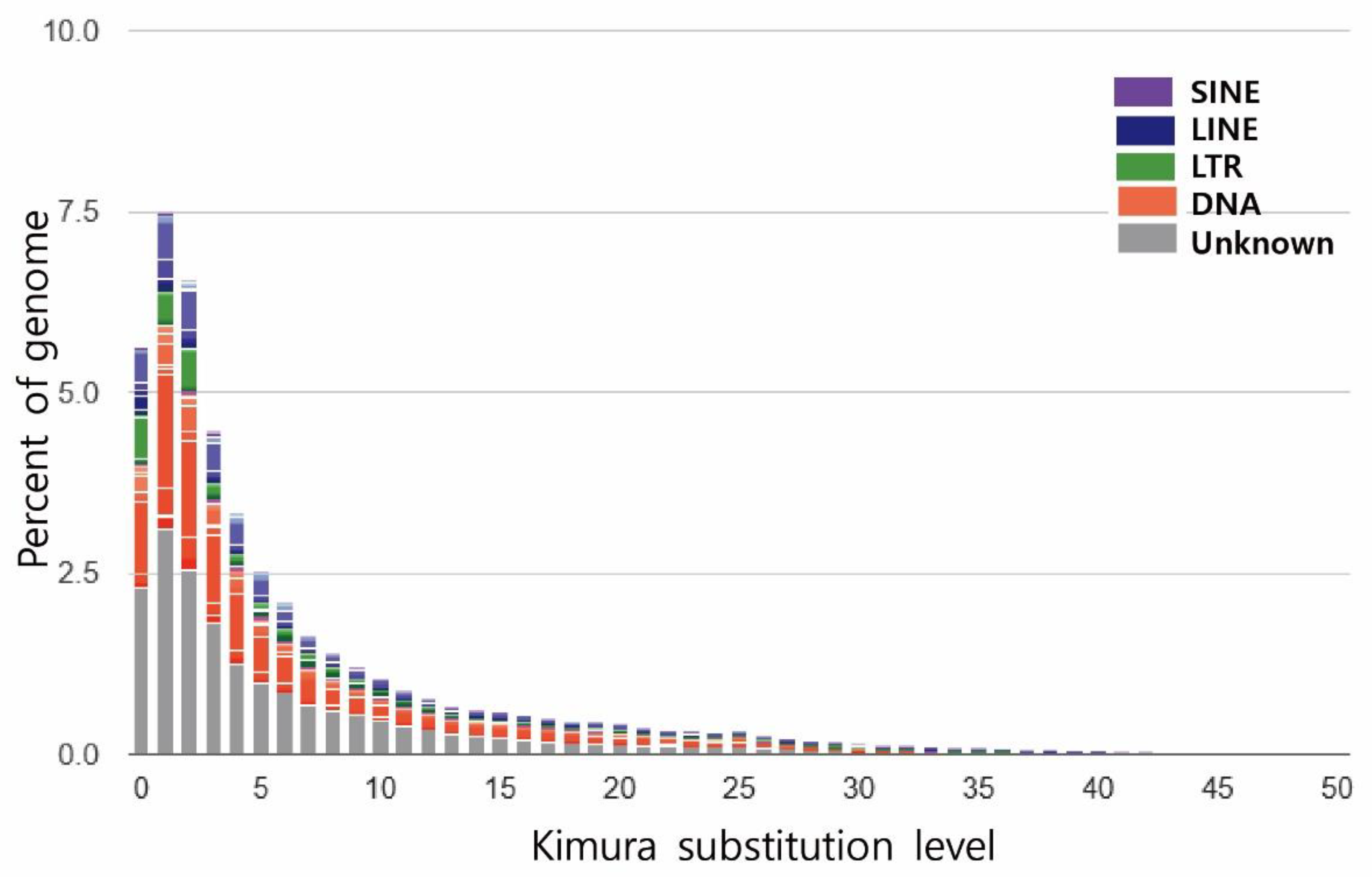
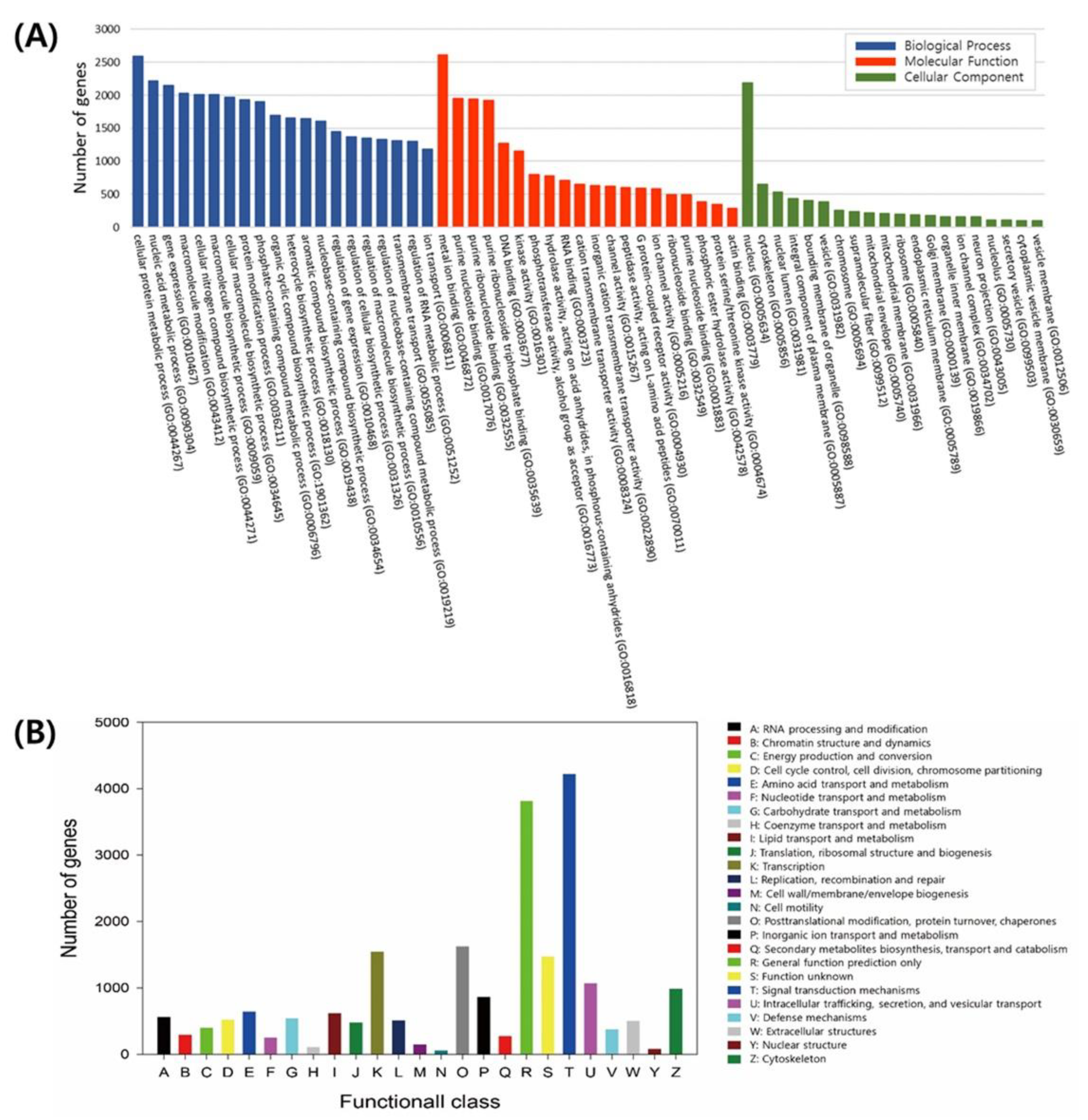
3.2. Genome Annotation
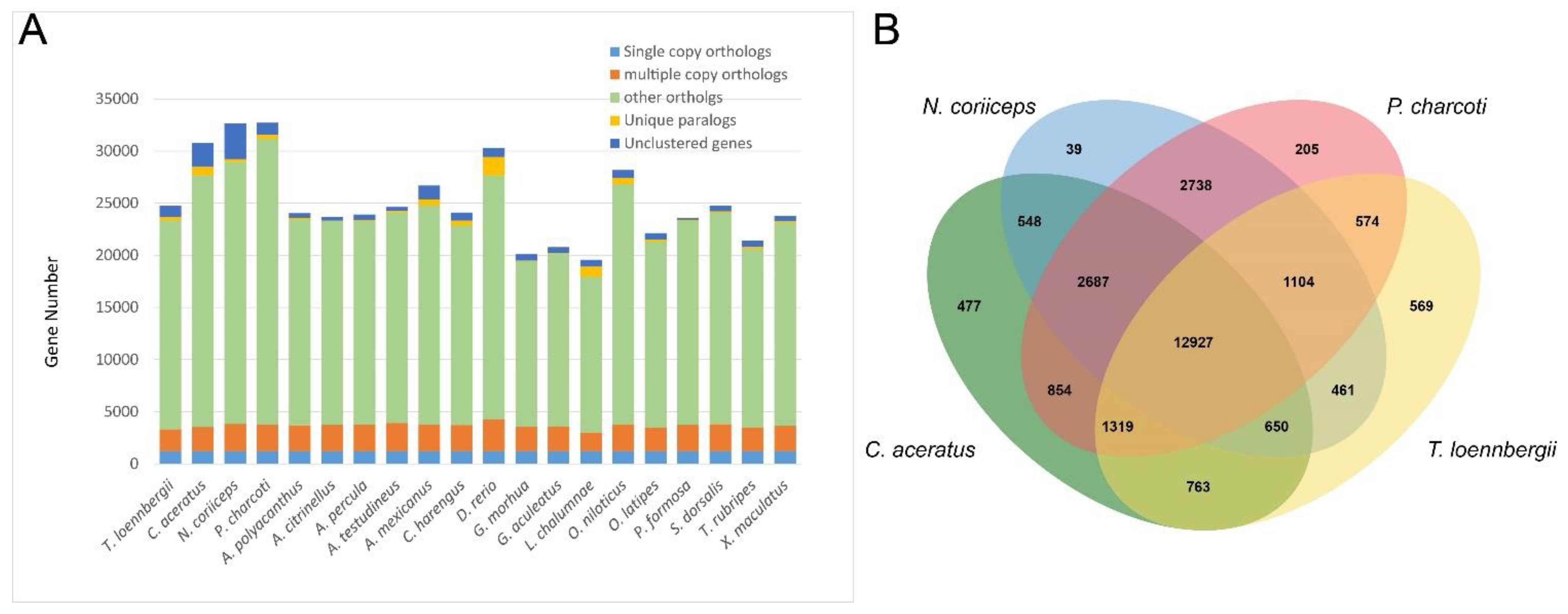
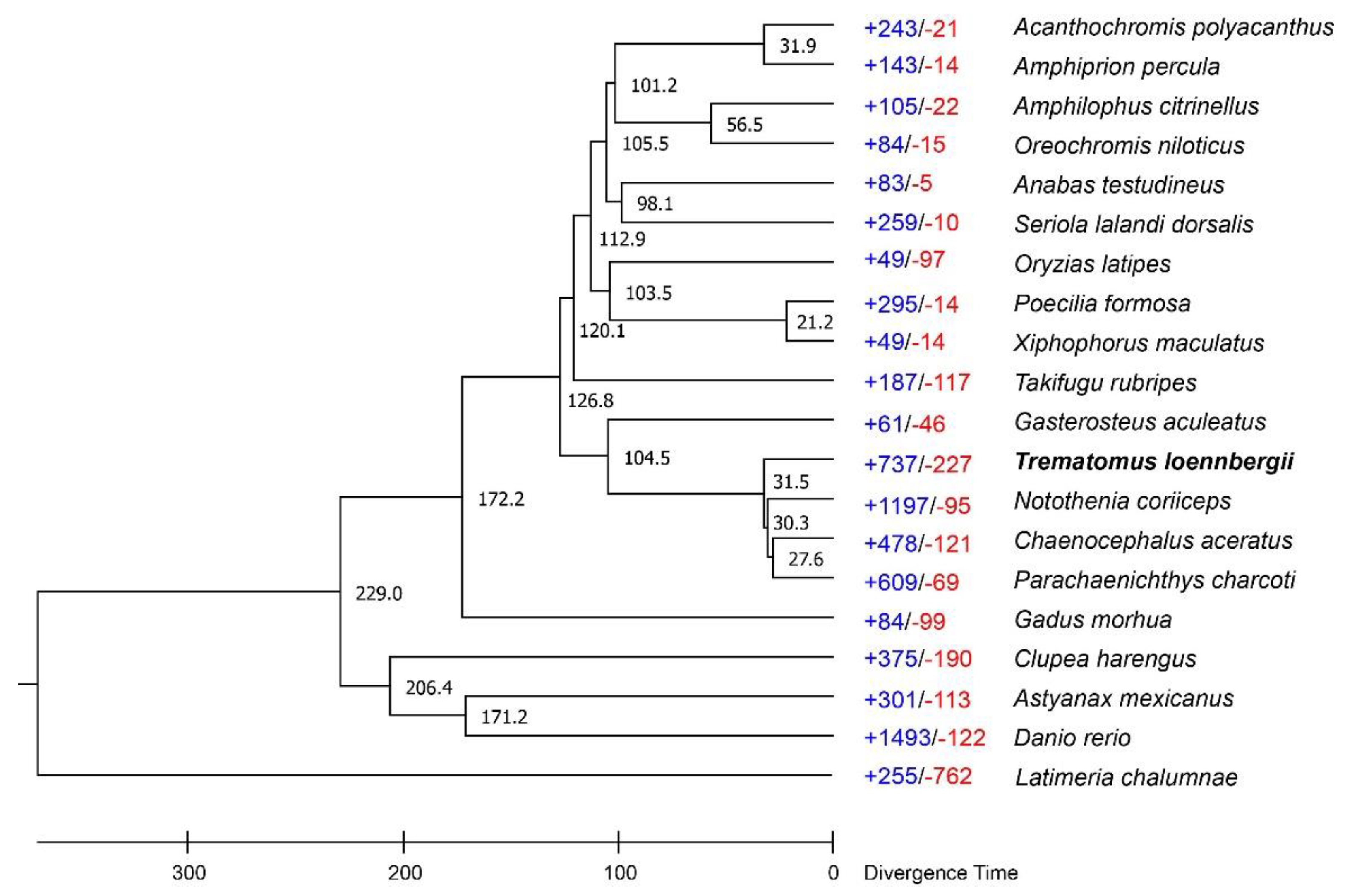
3.3. Gene Family Identification and Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beers, J.M.; Jayasundara, N. Antarctic notothenioid fish: What are the future consequences of ‘losses’ and ‘gains’ acquired during long-term evolution at cold and stable temperatures? J. Exp. Biol. 2015, 218, 1834–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVries, A.L.; Wohlschlag, D.E. Freezing resistance in some Antarctic fishes. Science 1969, 163, 1073–1075. [Google Scholar] [CrossRef]
- Buckley, B.A.; Place, S.P.; Hofmann, G.E. Regulation of heat shock genes in isolated hepatocytes from an Antarctic fish, Trematomus bernacchii. J. Exp. Biol. 2004, 207, 3649–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, G.E.; Buckley, B.A.; Airaksinen, S.; Keen, J.E.; Somero, G.N. Heat-shock protein expression is absent in the Antarctic fish Trematomus bernacchii (family Nototheniidae). J. Exp. Biol. 2000, 203, 2331–2339. [Google Scholar] [CrossRef]
- Kim, B.-M.; Amores, A.; Kang, S.; Ahn, D.-H.; Kim, J.-H.; Kim, I.-C.; Lee, J.H.; Lee, S.G.; Lee, H.; Lee, J. Antarctic Blackfin icefish genome reveals adaptations to extreme environments. Nat. Ecol. Evol. 2019, 3, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kock, K.-H. Antarctic Fish and Fisheries; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- Froese, R.; Pauly, D. FishBase. Available online: www.fishbase.org (accessed on 10 February 2020).
- De Witt, H.; Heemstra, P.; Gon, O. Nototheniidae; J. L. B. Smith Institute of Ichthyology: Grahamstown, South Africa, 1990; pp. 279–331. [Google Scholar]
- Miller, R.G. History and Atlas of the Fishes of the Antarctic Ocean; Foresta Inst: Carson City, NV, USA, 1993. [Google Scholar]
- Lautredou, A.-C.; Bonillo, C.; Denys, G.; Cruaud, C.; Ozouf-Costaz, C.; Lecointre, G.; Dettai, A. Molecular taxonomy and identification within the Antarctic genus Trematomus (Notothenioidei, Teleostei): How valuable is barcoding with COI? Polar Sci. 2010, 4, 333–352. [Google Scholar] [CrossRef]
- Dettaï, A.; Adamowizc, S.J.; Allcock, L.; Arango, C.P.; Barnes, D.K.; Barratt, I.; Chenuil, A.; Couloux, A.; Cruaud, C.; David, B. DNA barcoding and molecular systematics of the benthic and demersal organisms of the CEAMARC survey. Polar Sci. 2011, 5, 298–312. [Google Scholar] [CrossRef]
- Dettai, A.; Berkani, M.; Lautredou, A.-C.; Couloux, A.; Lecointre, G.; Ozouf-Costaz, C.; Gallut, C. Tracking the elusive monophyly of nototheniid fishes (Teleostei) with multiple mitochondrial and nuclear markers. Mar. Genom. 2012, 8, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Steinke, D.; Dettai, A.; McMillan, P.; Welsford, D.; Stewart, A.; Ward, R. DNA barcodes and species identifications in Ross Sea and Southern Ocean fishes. Polar Biol. 2012, 35, 1297–1310. [Google Scholar] [CrossRef]
- Ghigliotti, L.; Cheng, C.C.-H.; Ozouf-Costaz, C.; Vacchi, M.; Pisano, E. Cytogenetic diversity of notothenioid fish from the Ross sea: Historical overview and updates. Hydrobiologia 2015, 761, 373–396. [Google Scholar] [CrossRef]
- Auvinet, J.; Graça, P.; Belkadi, L.; Petit, L.; Bonnivard, E.; Dettaï, A.; Detrich, W.; Ozouf-Costaz, C.; Higuet, D. Mobilization of retrotransposons as a cause of chromosomal diversification and rapid speciation: The case for the Antarctic teleost genus Trematomus. BMC Genom. 2018, 19, 339. [Google Scholar] [CrossRef] [Green Version]
- Pisano, E.; Ozouf-Costaz, C. Chromosome change and the evolution in the Antarctic fish suborder Notothenioidei. Antarct. Sci. 2000, 12, 334–342. [Google Scholar] [CrossRef]
- Amores, A.; Wilson, C.A.; Allard, C.A.; Detrich, H.W.; Postlethwait, J.H. Cold fusion: Massive karyotype evolution in the Antarctic Bullhead Notothen Notothenia coriiceps. G3 Genes Genomes Genet. 2017, 7, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Ozouf-Costaz, C.; Hureau, J.; Beaunier, M. Chromosome studies on fish of the suborder Notothenioidei collected in the Weddell Sea during EPOS 3 cruise. Cybium 1991, 15, 271–289. [Google Scholar]
- Ozouf-Costaz, C.; Pisano, E.; Thaeron, C.; Hureau, J. Karyological survey of the Notothenioid fish occurring in Adélie Land (Antarctica). In Proceedings of the 5th Indo-Pac Fish Conf Nouméa, Nouméa, New Caledonia, 3–8 November 1997; pp. 427–440. [Google Scholar]
- Morescalchi, A.; Pisano, E.; Stanyon, R.; Morescalchi, M. Cytotaxonomy of antarctic teleosts of the Pagothenia/Trematomus complex (Nototheniidae, Perciformes). Polar Biol. 1992, 12, 553–558. [Google Scholar] [CrossRef]
- Giani, A.M.; Gallo, G.R.; Gianfranceschi, L.; Formenti, G. Long walk to genomics: History and current approaches to genome sequencing and assembly. Comput. Struct. Biotechnol. J. 2020, 18, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, N.; Dekker, J. High-throughput genome scaffolding from in vivo DNA interaction frequency. Nat. Biotechnol. 2013, 31, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, L.; Huang, H.; Zhao, M.; Jiang, K.; Zhang, F.; Zhao, M.; Chen, X.; Ma, L. The complete mitochondrial genome sequence and gene organization of Trematomus bernacchii (Perciformes: Nototheniidae) with phylogenetic consideration. Mitochondrial DNA Part B 2016, 1, 50–51. [Google Scholar] [CrossRef]
- Alam, M.J.; Kim, J.-H.; Andriyono, S.; Lee, J.-H.; Lee, S.R.; Park, H.; Kim, H.-W. Characterization of complete mitochondrial genome and gene organization of sharp-spined notothenia, Trematomus pennellii (Perciformes: Nototheniidae). Mitochondrial DNA Part B 2019, 4, 648–649. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.; Im, T.-E.; Lee, S.J.; Jo, E.; Kim, J.; Kim, S.H.; Chi, Y.M.; Kim, J.-H.; Park, H. The complete mitochondrial genome of Trematomus loennbergii (Perciformes, Nototheniidae). Mitochondrial DNA Part B 2021, 6, 1032–1033. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef]
- Belton, J.-M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Soderlund, C.; Bomhoff, M.; Nelson, W.M. SyMAP v3. 4: A turnkey synteny system with application to plant genomes. Nucleic Acids Res. 2011, 39, e68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef] [Green Version]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2010, 39, D225–D229. [Google Scholar] [CrossRef] [Green Version]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.-C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Eddy, S.R.; Mitchison, G.; Durbin, R. Maximum discrimination hidden Markov models of sequence consensus. J. Comput. Biol. 1995, 2, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, P.P.; Daub, J.; Tate, J.; Moore, B.L.; Osuch, I.H.; Griffiths-Jones, S.; Finn, R.D.; Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 2010, 39, D141–D145. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.B.; Dudley, J.; Kumar, S. TimeTree: A public knowledge-base of divergence times among organisms. Bioinformatics 2006, 22, 2971–2972. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.V.; Thomas, G.W.; Lugo-Martinez, J.; Hahn, M.W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 2013, 30, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Deng, X.; Wang, J.; Li, Y.; Wu, S.; Yang, S.; Chao, J.; Chen, Y.; Zhang, S.; Shi, M.; Tian, W. Comparative transcriptome analysis reveals phytohormone signalings, heat shock module and ROS scavenger mediate the cold-tolerance of rubber tree. Sci. Rep. 2018, 8, 4931. [Google Scholar] [CrossRef]
- Li, B.; Ning, L.; Zhang, J.; Bao, M.; Zhang, W. Transcriptional profiling of Petunia seedlings reveals candidate regulators of the cold stress response. Front. Plant Sci. 2015, 6, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zieger, M.A.; Gupta, M.P.; Wang, M. Proteomic analysis of endothelial cold-adaptation. BMC Genom. 2011, 12, 630. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly | Contigs | Scaffolds |
|---|---|---|
| Number | 1482 | 1132 |
| Total size of assembly (bp) | 944,447,341 | 944,482,341 |
| Longest contig (bp) | 17,998,618 | 48,466,630 |
| N50 contigs length (bp) | 1,726,674 | 24,660,741 |
| Number of scaffolds > 9 Mb | 4 | 23 |
| Actinopterygii_odb10 | Number | Percentage (%) |
|---|---|---|
| Complete BUSCOs (C) | 3291 | 90.4 |
| Complete and single-copy BUSCOs (S) | 3222 | 88.5 |
| Complete and duplicated BUSCOs (D) | 69 | 1.9 |
| Fragmented BUSCOs (F) | 96 | 2.6 |
| Missing BUSCOs (M) | 253 | 7.0 |
| Total BUSCO groups searched | 3640 | - |
| Annotation Database | Annotated Number | Percentage (%) |
|---|---|---|
| No. Genes | 24,525 | - |
| nr Annotation | 23,016 | 93.8 |
| GO Annotation | 19,389 | 79.1 |
| KEGG Annotation | 11,546 | 47.1 |
| KOG Annotation | 15,801 | 64.4 |
| Pfam Annotation | 17,313 | 70.6 |
| Swissprot Annotation | 19,809 | 80.8 |
| TrEMBL Annotation | 22,932 | 93.5 |
| - | Count | Length Sum (bp) |
| Exon | 329,468 | 62,037,587 |
| CDS | 311,918 | 48,174,219 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, E.; Lee, S.J.; Kim, J.-H.; Parker, S.J.; Choi, E.; Kim, J.; Han, S.-R.; Oh, T.-J.; Park, H. Chromosomal-Level Assembly of Antarctic Scaly Rockcod, Trematomus loennbergii Genome Using Long-Read Sequencing and Chromosome Conformation Capture (Hi-C) Technologies. Diversity 2021, 13, 668. https://doi.org/10.3390/d13120668
Jo E, Lee SJ, Kim J-H, Parker SJ, Choi E, Kim J, Han S-R, Oh T-J, Park H. Chromosomal-Level Assembly of Antarctic Scaly Rockcod, Trematomus loennbergii Genome Using Long-Read Sequencing and Chromosome Conformation Capture (Hi-C) Technologies. Diversity. 2021; 13(12):668. https://doi.org/10.3390/d13120668
Chicago/Turabian StyleJo, Euna, Seung Jae Lee, Jeong-Hoon Kim, Steven J. Parker, Eunkyung Choi, Jinmu Kim, So-Ra Han, Tae-Jin Oh, and Hyun Park. 2021. "Chromosomal-Level Assembly of Antarctic Scaly Rockcod, Trematomus loennbergii Genome Using Long-Read Sequencing and Chromosome Conformation Capture (Hi-C) Technologies" Diversity 13, no. 12: 668. https://doi.org/10.3390/d13120668
APA StyleJo, E., Lee, S. J., Kim, J. -H., Parker, S. J., Choi, E., Kim, J., Han, S. -R., Oh, T. -J., & Park, H. (2021). Chromosomal-Level Assembly of Antarctic Scaly Rockcod, Trematomus loennbergii Genome Using Long-Read Sequencing and Chromosome Conformation Capture (Hi-C) Technologies. Diversity, 13(12), 668. https://doi.org/10.3390/d13120668