
Implications of Historical and Contemporary Processes on Genetic Differentiation of a Declining Boreal Songbird: The Rusty Blackbird

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. ddRAD-seq Library Preparation
2.3. Bioinformatics
2.4. mtDNA Sequencing
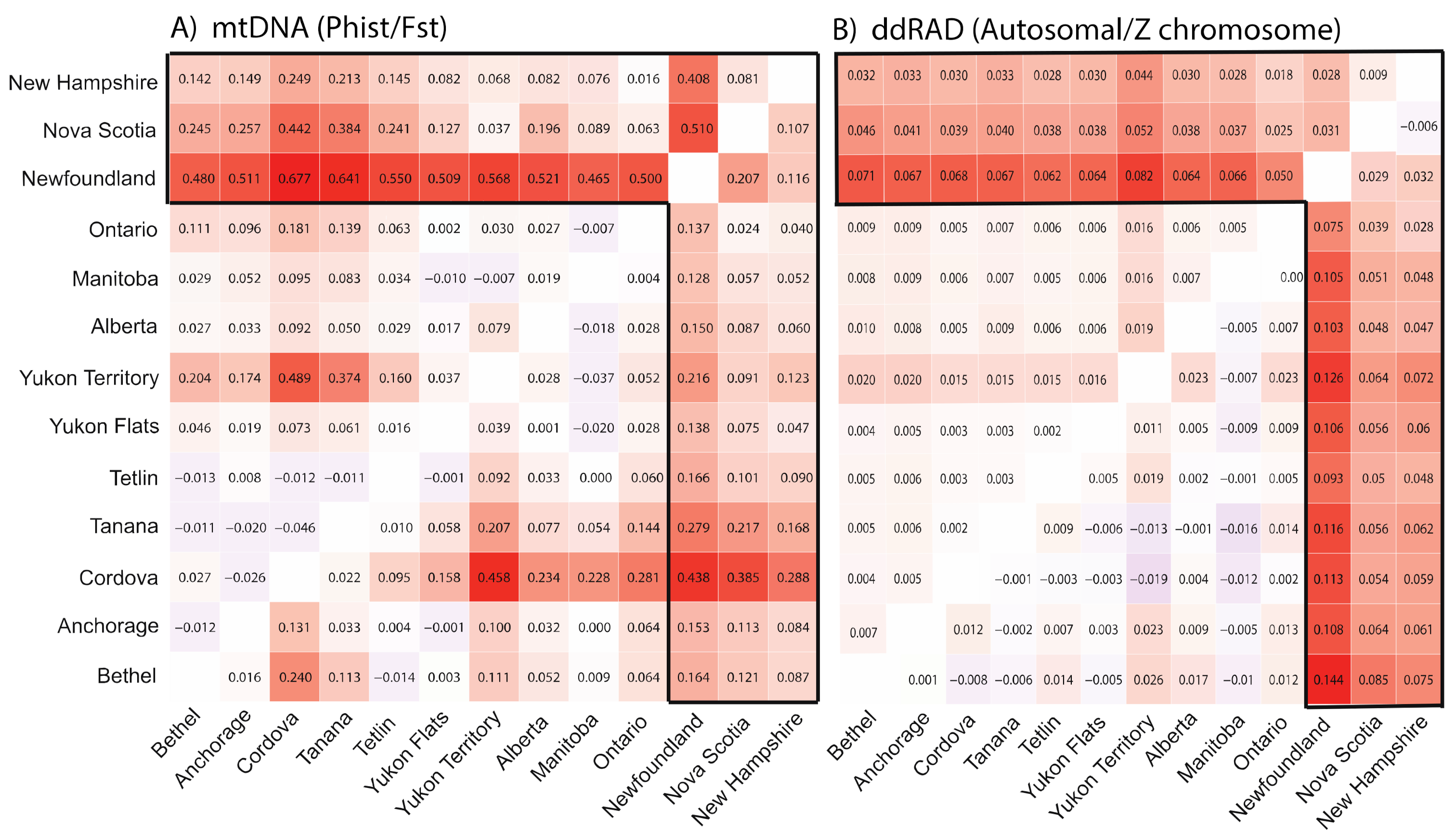
2.5. Population Divergence and Nucleotide Diversity
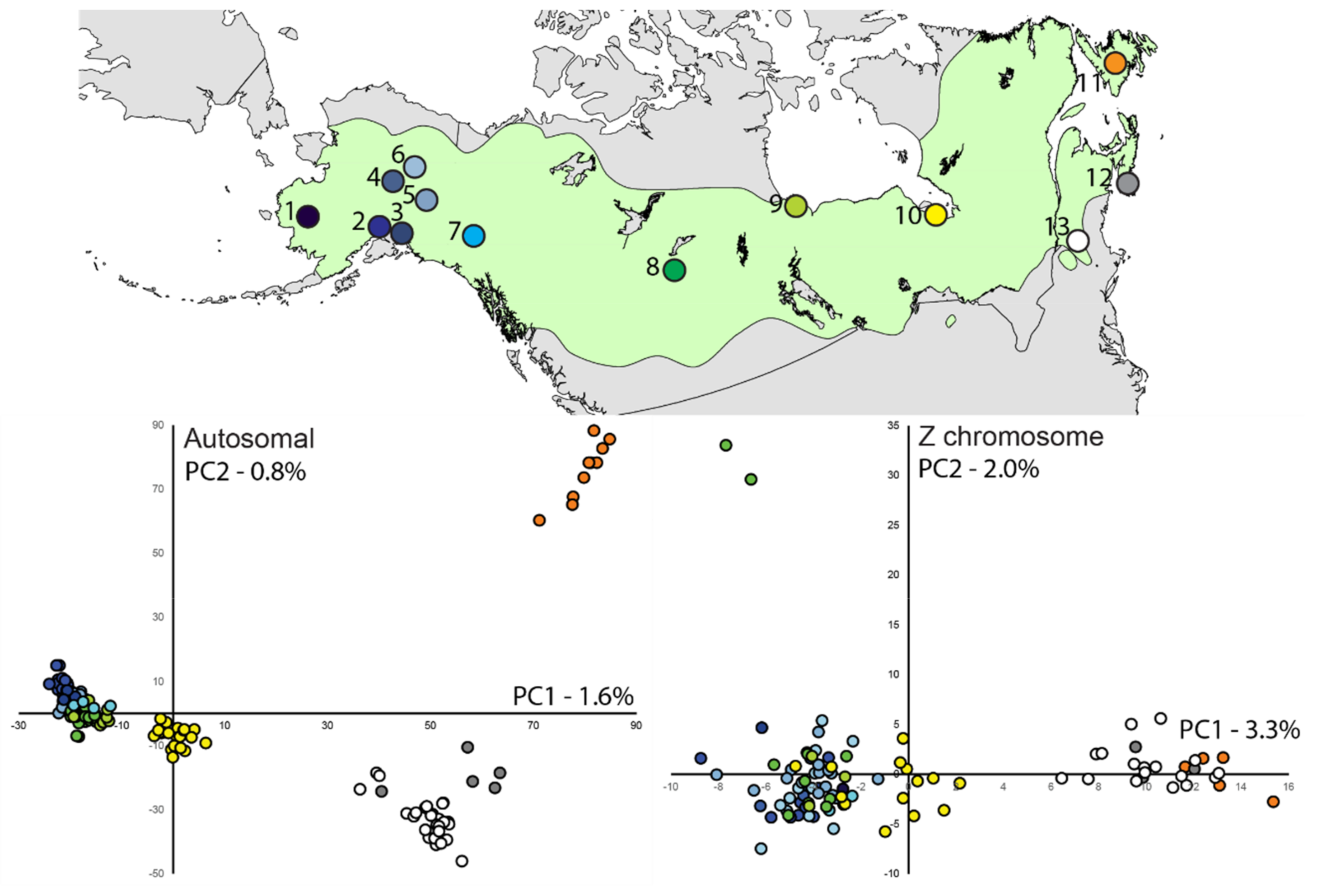
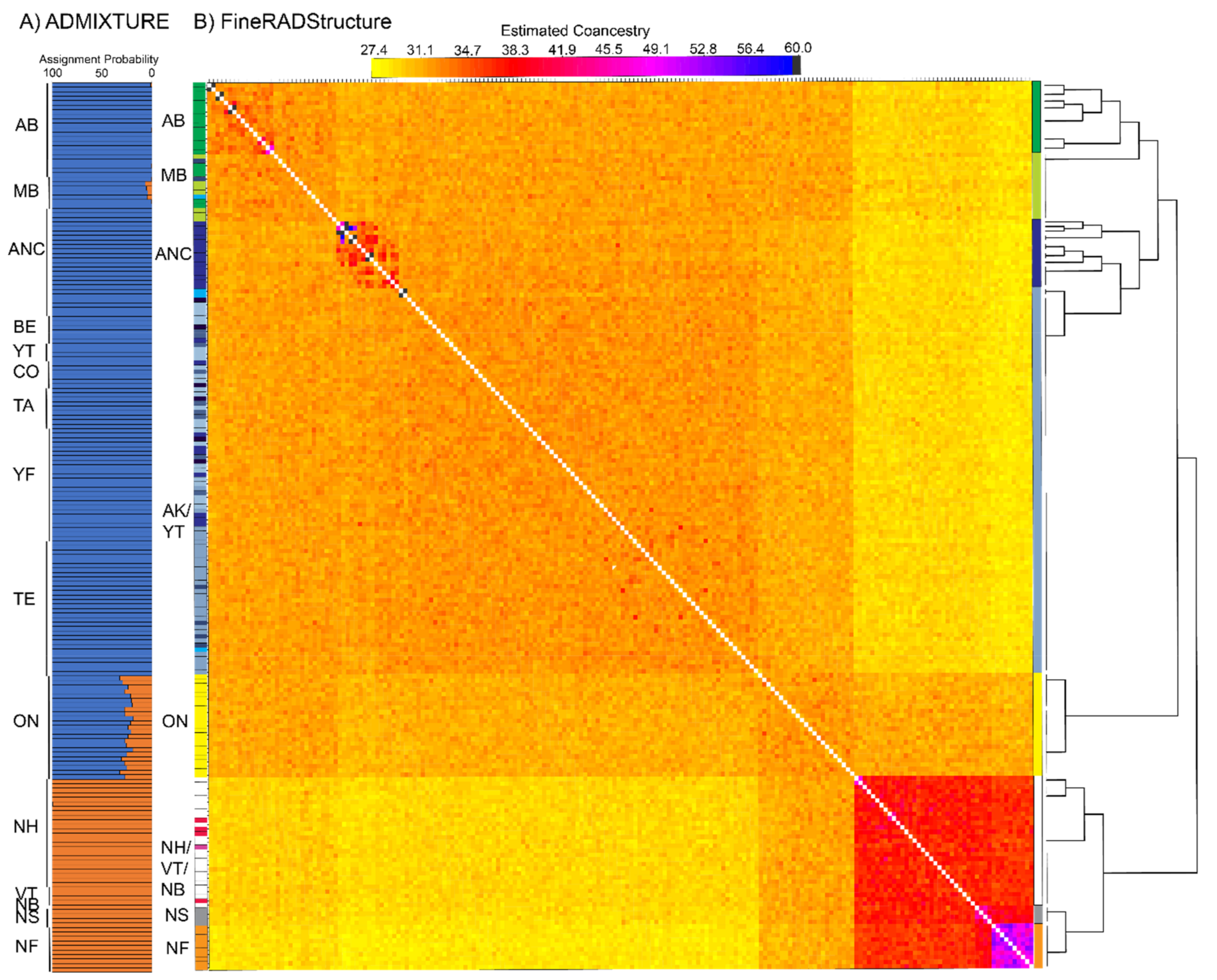
2.6. Population Structure—ddRAD
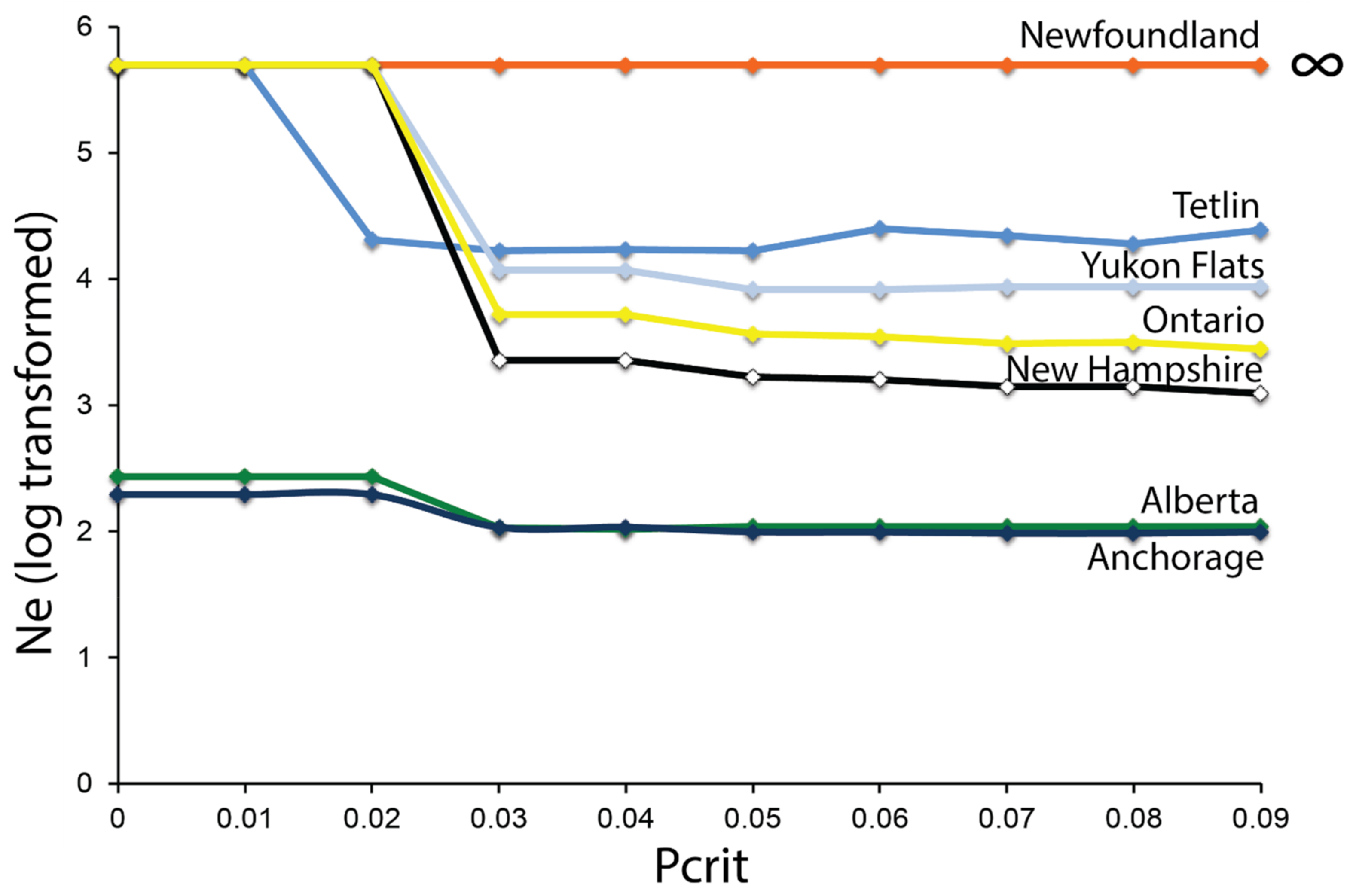
2.7. Effective Population Size—ddRAD
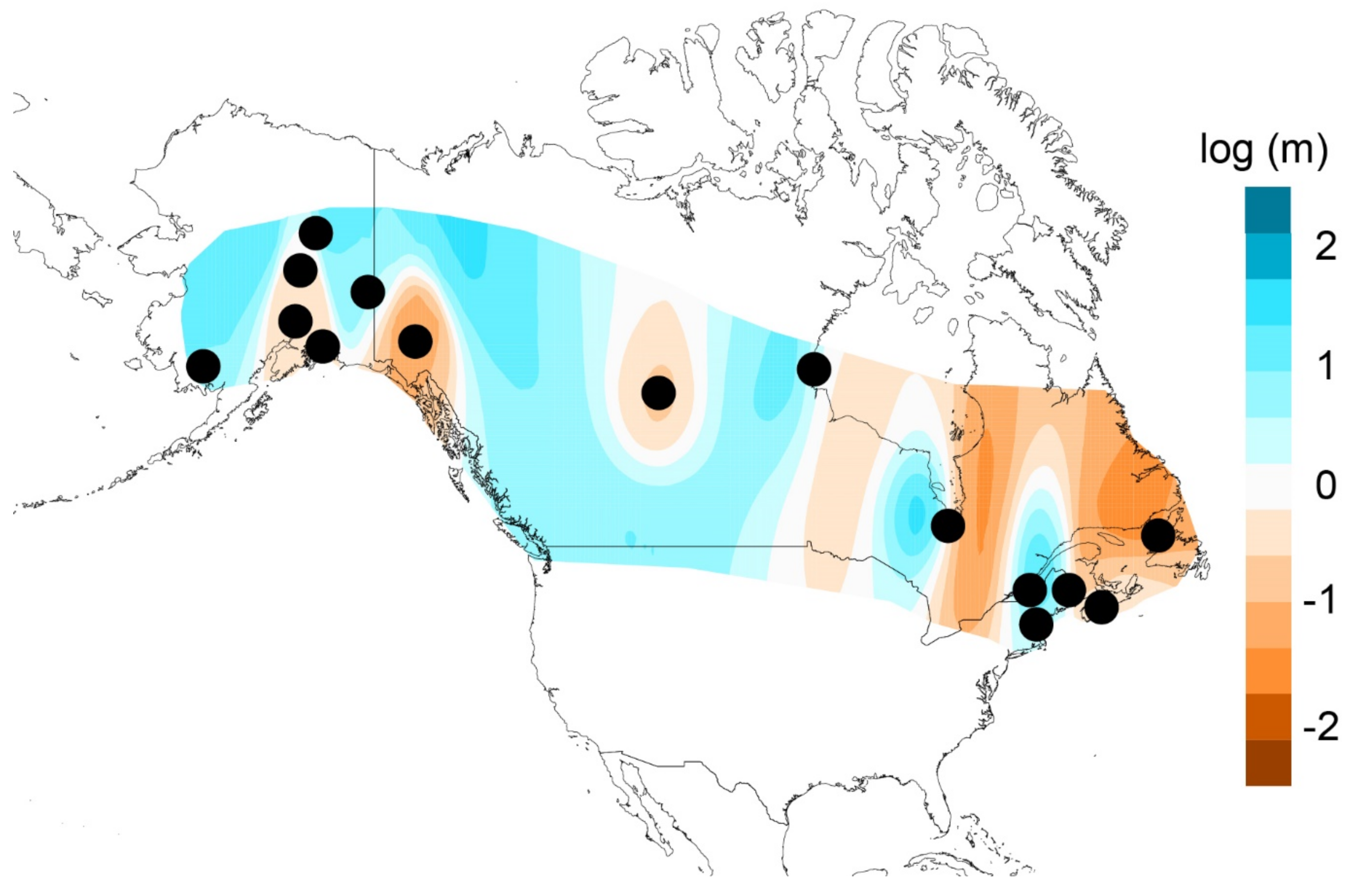
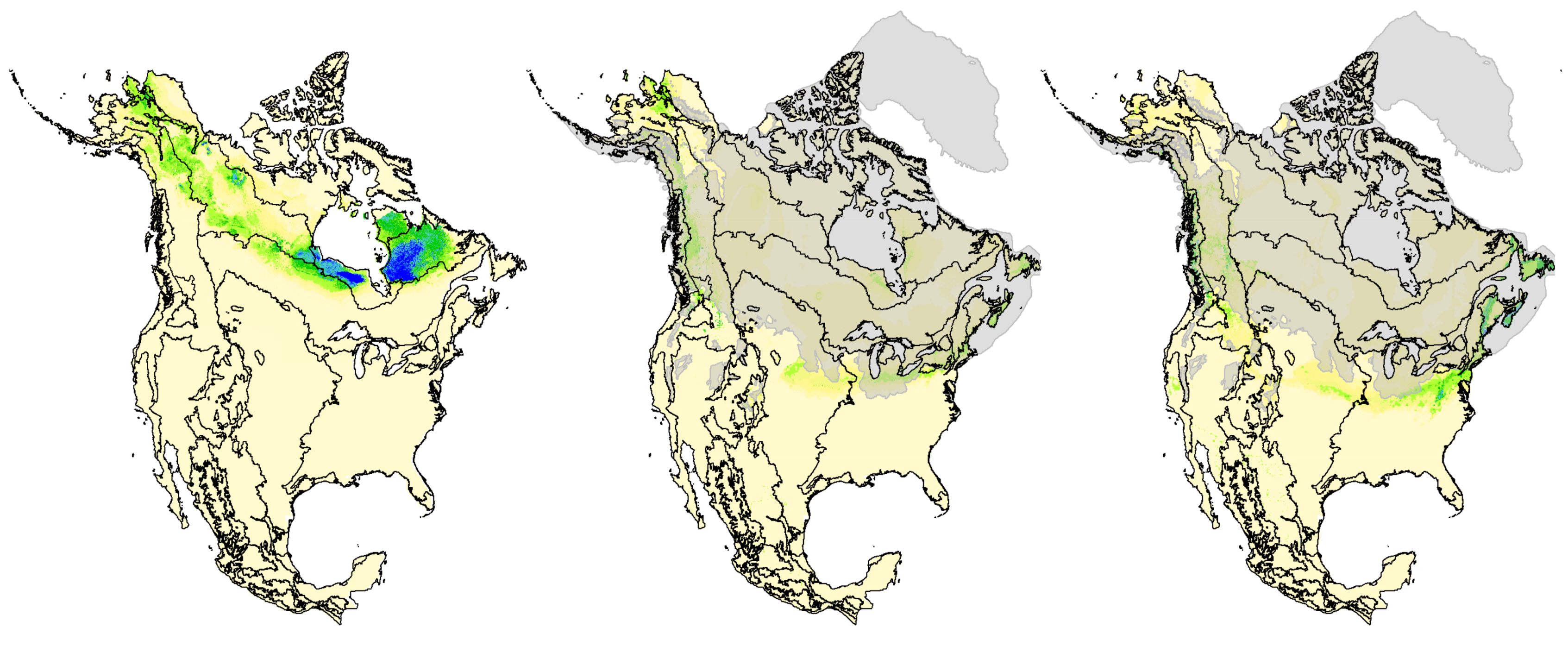
2.8. Hindcasted Paleo-Distributions of Breeding Rusty Blackbirds
3. Results
3.1. Bioinformatics—ddRAD
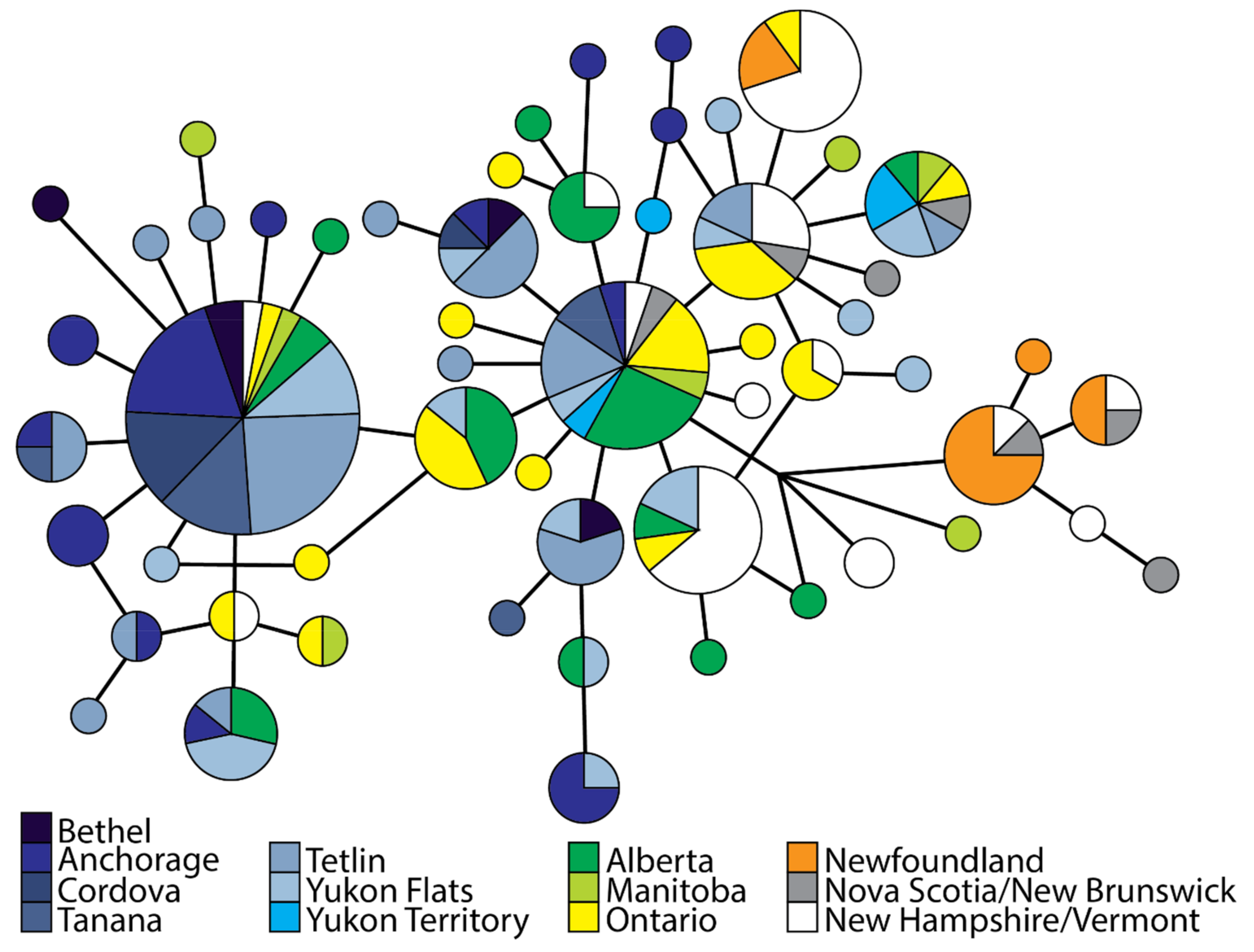
3.2. Population Divergence and Molecular Diversity
3.3. Population Structure—ddRAD
3.4. Effective Population Size—ddRAD
3.5. Hindcasted Paleo-Distributions of Breeding Rusty Blackbirds
4. Discussion
4.1. Pleistocene Influences on Patterns of Genomic Diversity
4.2. Isolation in Newfoundland
4.3. Contemporary Influences on Patterns of Genomic Variation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, A.R.; Wiens, J.A.; Milne, B.T.; Crist, T.O. Animal movements and population dynamics in heterogeneous landscapes. Landsc. Ecol. 1992, 7, 63–75. [Google Scholar] [CrossRef]
- Robbins, A.M.; Robbins, M.M. Fitness consequences of dispersal decisions for male mountain gorillas (Gorilla beringei beringei). Behav. Ecol. Sociobiol. 2005, 58, 295–309. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Keyghobadi, N. The genetic implications of habitat fragmentation for animals. Can. J. Zool. 2007, 85, 1049–1064. [Google Scholar] [CrossRef]
- Radespiel, U.; Bruford, M.W. Fragmentation genetics of rainforests animals: Insights from recent studies. Conserv. Genet 2014, 15, 245–260. [Google Scholar] [CrossRef]
- Oretgo, J.; Aguiree, M.P.; Noguerales, V.; Cordero, P.J. Consequences of extensive habitat fragmentation in landscape-level patterns of genetic diversity and structure in the Mediterranean esparto grasshopper. Evol. Appl. 2015, 8, 621–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Villemeruil, P.; Rutschmann, A.; Lee, K.D.; Ewen, J.G.; Brekke, P.; Santure, A.W. Little adaptive potential in a threatened passerine bird. Curr. Biol. 2019, 29, 889–894. [Google Scholar] [CrossRef] [Green Version]
- Ouborg, N.J.; Vergeer, P.; Mix, C. The rough edges of the conservation genetics paradigm for plants. J. Ecol. 2006, 94, 1233–1248. [Google Scholar] [CrossRef]
- Leonardi, S.; Piovani, P.; Scalfi, M.; Piotti, A.; Giannini, R.; Menozzi, P. Effect of habitat fragmentation on the genetic diversity and structure of peripheral populations of beech in central Italy. J. Hered. 2012, 103, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, K.; Harrisson, K.A.; Hogan, F.E.; Cooke, R.; Clarke, R.H. Reduced gene flow in a vulnerable species reflects two centuries of habitat loss and fragmentation. Ecosphere 2018, 9, e02114. [Google Scholar] [CrossRef] [Green Version]
- Ewers, R.M.; Didham, R.K. Confounding factors in the detection of species responses to habitat fragmentation. Biol. Rev. 2006, 81, 117–142. [Google Scholar] [CrossRef] [PubMed]
- Amos, J.N.; Bennett, A.F.; Mac Nally, R.; Newell, G.; Pavlova, A.; Radford, J.Q.; Thomson, J.R.; White, M.; Sunnucks, P. Predicting landscape-genetic consequences of habitat loss, fragmentation and mobility for multiple species of woodland birds. PLoS ONE 2012, 7, e30888. [Google Scholar] [CrossRef] [Green Version]
- Dharmarajan, G.; Beasley, J.C.; Fike, J.A.; Rhodes, O.E., Jr. Effects of landscape, demographic and behavioral factors on kin structure: Testing ecological predictions in a mesopredator with high dispersal capability. Anim. Conserv. 2014, 17, 225–234. [Google Scholar] [CrossRef]
- Lindsay, D.L.; Barr, K.R.; Lance, R.F.; Tweddale, S.A.; Hayden, T.J.; Leberg, P.L. Habitat fragmentation and genetic diversity of an endangered, migratory songbird, the golden-cheeked warbler (Dendroica chrysoparia). Mol. Ecol. 2008, 17, 2122–2133. [Google Scholar] [CrossRef]
- Ruegg, K.C.; Anderson, E.C.; Paxton, K.L.; Apkenas, V.; Lao, S.; Siegel, R.B.; DeSante, D.F.; Moore, F.; Smith, T.B. Mapping migration in a songbird using high-resolution genetic markers. Mol. Ecol. 2014, 23, 5726–5739. [Google Scholar] [CrossRef]
- Robertson, E.P.; Fletcher, R.J., Jr.; Cattau, C.E.; Udell, B.J.; Reichert, B.E.; Austin, J.D.; Valle, D. Isolating the roles of movement and reproduction on effective connectivity alters conservation priorities for an endangered bird. Proc. Natl. Acad. Sci. USA 2018, 115, 8591–8596. [Google Scholar] [CrossRef] [Green Version]
- Robertson, E.P.; Fletcher, R.J., Jr.; Austin, J.D.; Valle, D. The number of breeders explains genetic connectivity in an endangered bird. Mol. Ecol. 2019, 28, 2746–2756. [Google Scholar] [PubMed]
- Coulon, A.; Fitzpatrick, J.W.; Bowman, R.; Lovette, J.J. Effects of habitat fragmentation on effective dispersal of Florida scrub jays. Conserv. Biol. 2019, 24, 1080–1088. [Google Scholar] [CrossRef]
- Schindler, D.W.; Lee, P.G. Comprehensive conservation planning to protect biodiversity and ecosystem services in Canadian boreal regions under a warming climate and increasing exploitation. Biol. Conserv. 2010, 143, 1571–1586. [Google Scholar] [CrossRef]
- Wells, J.V. Chapter 1: Threats and conservation status. In Boreal Birds of North America: A Hemispheric View of Their Conservation Links and Significance; Studies in Avian Biology; Wells, J.V., Ed.; University of California Press: Berkeley, CA, USA, 2011; Volume 41, pp. 1–6. [Google Scholar]
- Wells, J.V.; Blancher, P.J. Global role for sustaining bird populations. In Boreal Birds of North America: A Hemispheric View of Their Conservation Links and Significance; Studies in Avian Biology; Wells, J.V., Ed.; University of California Press: Berkeley, CA, USA, 2011; Volume 41, pp. 7–22. [Google Scholar]
- Wells, J.; Stralberg, D.; Childs, D. Boreal Forest Refuge: Conserving North America’s Bird Nursery in the Face of Climate Change; Boreal Songbird Initiative: Seattle, WA, USA, 2018. [Google Scholar]
- Rosenberg, K.V.; Dokter, A.M.; Blancher, P.J.; Sauer, J.R.; Smith, A.C.; Smith, P.A.; Stanton, J.C.; Panjabi, A.; Helft, L.; Parr, M.; et al. Decline of the North American avifauna. Science 2019, 366, 120–124. [Google Scholar] [CrossRef]
- Soykan, C.U.; Sauer, J.; Schuetz, J.G.; LeBaron, G.S.; Dale, K.; Langham, G.M. Population trends for North American winter birds based on hierarchical models. Ecosphere 2016, 7, e01351. [Google Scholar] [CrossRef]
- Greenberg, R.; Droege, S. On the decline of the rusty blackbird and the use of ornithological literature to document long-term population trends. Conserv. Biol. 1999, 13, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, R.; Matsuoka, S.M. Rusty blackbird: Mysteries of a species in decline (Euphagus carolinus). Condor 2010, 112, 770–777. [Google Scholar] [CrossRef]
- Greenberg, R.; Demarest, D.W.; Matsuoka, S.M.; Mettke-Hofmann, C.; Evers, D.; Hamel, P.B.; Luscier, J.; Powell, L.L.; Shaw, D.; Avery, M.L.; et al. Understanding declines in rusty blackbirds. In Boreal Birds of North America: A Hemispheric View of Their Conservation Links and Significance; Studies in Avian Biology; Wells, J.V., Ed.; University of California Press: Berkeley, CA, USA, 2011; Volume 41, pp. 107–126. [Google Scholar]
- Edmonds, S.T.; Evers, D.C.; Cristol, D.A.; Mettke-Hofmann, C.; Powell, L.L.; McGann, A.J.; Armiger, J.W.; Lane, O.P.; Tessler, D.F.; Newell, P.; et al. Geographic and seasonal variation in mercury exposure of the declining rusty blackbird. Condor 2010, 112, 789–799. [Google Scholar] [CrossRef]
- Edmonds, S.T.; O’Driscoll, N.J.; Hillier, N.K.; Atwood, J.L.; Evers, D.C. Factors regulating the bioavailability of methylmercury to breeding rusty blackbirds in northeastern wetlands. Environ. Pollut. 2012, 171, 148–154. [Google Scholar] [CrossRef]
- Perkins, M.; Lane, O.P.; Evers, D.C.; Sauer, A.; Adams, E.M.; O’Driscoll, N.J.; Edmunds, S.T.; Jackson, A.K.; Hagelin, J.C.; Trimble, J.; et al. Historical patterns in mercury exposure for North American songbirds. Ecotoxicology 2020, 29, 1161–1173. [Google Scholar] [CrossRef]
- Wright, J.R.; Powell, L.L.; Tonra, C.M. Automated telemetry reveals staging behavior in a declining migratory passerine. Auk Ornithol. Adv. 2018, 135, 461–476. [Google Scholar] [CrossRef] [Green Version]
- Powell, L.L.; Hodgman, T.P.; Glanz, W.E.; Osenton, J.D.; Fisher, C.M. Nest-site selection and nest survival of the rusty blackbird: Does timber management adjacent to wetlands create ecological traps? Condor 2010, 112, 800–809. [Google Scholar] [CrossRef]
- McClure, C.J.W.; Rolek, B.W.; McDonald, K.; Hill, G.E. Climate change and the decline of a once common bird. Ecol. Evol. 2012, 2, 370–378. [Google Scholar] [CrossRef]
- Hamel, P.B.; De Stevens, D.; Leininger, T.; Wilson, R. Historical trends in rusty blackbird nonbreeding habitat in forested wetlands. In Proceedings of the 4th International Partners in Flight Conference, McAllen, TX, USA, 13–16 February 2008; Rich, T., Arizmendi, C., Thompson, C., Demarest, D., Eds.; Partners in Flight, 2009; pp. 341–353. [Google Scholar]
- Hobson, K.A.; Greenberg, R.; Van Wilgenburg, S.L.; Mettke-Hofmann, C. Migratory connectivity in the rusty blackbird: Isotopic evidence from feathers of historical and contemporary specimens. Condor 2010, 112, 778–788. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.A.; Matsuoka, S.M.; Tessler, D.F.; Greenberg, R.; Fox, J.W. Identifying migratory pathways used by rusty blackbirds breeding in southcentral Alaska. Wilson J. Ornithol. 2012, 124, 698–703. [Google Scholar] [CrossRef]
- Burleigh, T.D.; Peters, H.S. Geographic variation in Newfoundland birds. Proc. Biol. Soc. Wash. 1948, 61, 111–126. [Google Scholar]
- Dyke, A.S. Late Quaternary vegetation history of northern North America based on pollen, macrofossil, and faunal remains. Geogr. Phys. Quatern. 2005, 59, 211–262. [Google Scholar]
- Shafer, A.B.A.; Cullingham, C.I.; Cote, S.D.; Coltman, D.W. Of glaciers and refugia: A decade of study sheds new light on the phylogeography of northwestern North America. Mol. Ecol. 2010, 19, 4589–4621. [Google Scholar] [CrossRef]
- Sonsthagen, S.A.; Talbot, S.L.; Scribner, K.T.; McCracken, K.G. Multilocus phylogeography and population structure of common eiders breeding in North America and Scandinavia. J. Biogeogr. 2011, 38, 1368–1380. [Google Scholar] [CrossRef]
- van Els, P.; Cicero, C.; Klicka, J. High latitudes and high genetic diversity: Phylogeography of a widespread boreal bird, the gray jay (Perisoreus canadensis). Mol. Phylogenet. Evol. 2012, 63, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Lait, L.A.; Burg, T.M. When east meets west: Population structure of high-latitude resident species, the boreal chickadee (Poecile hudsonicus). Heredity 2013, 111, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, G.M. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. B Biol. Sci. 2004, 359, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Dohms, K.M.; Graham, B.A.; Burg, T.M. Multilocus genetic analysis and spatial modeling reveal complex population structure and history in a widespread resident North American passerine (Perisoreus canadensis). Ecol. Evol. 2017, 7, 9869–9889. [Google Scholar] [CrossRef] [Green Version]
- Colbeck, G.J.; Gibbs, H.L.; Marra, P.P.; Hobson, K.; Webster, M.S. Phylogeography of a widespread North American migratory songbird (Setophaga ruticilla). J. Hered. 2008, 99, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Ralston, J.; Kirchman, J.J. Continent-scale genetic structure in a boreal forest migrant, the blackpoll warbler (Setophaga striata). Auk 2012, 129, 467–478. [Google Scholar]
- Lovette, I.J. Glacial cycles and the tempo of avian speciation. Trends Ecol. Evol. 2005, 20, 57–59. [Google Scholar] [CrossRef]
- Zamudio, K.R.; Bell, R.C.; Mason, N.A. Phenotypes in phylogeography: Species’ traits, environmental variation, and vertebrate diversification. Proc. Natl. Acad. Sci. USA 2016, 113, 8041–8048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cote, J.; Bestion, E.; Jacob, S.; Travis, J.; Legrand, D.; Baguette, M. Evolution of dispersal strategies and dispersal syndromes in fragmented landscapes. Ecography 2017, 40, 56–73. [Google Scholar] [CrossRef]
- Cayuela, H.; Rougemont, Q.; Prunier, J.G.; Soore, J.-S.; Clobert, J.; Besnard, A.; Bernatchez, L. Demographic and genetic approaches to study dispersal in wild animal populations: A methodological review. Mol. Ecol. 2018, 27, 3976–4010. [Google Scholar] [CrossRef]
- Cornelius, C.; Awade, M.; Cândia-Gallardo, C.; Sieving, K.E.; Metzger, J.P. Habitat fragmentation drives inter-population variation in dispersal behavior in a Neotropical rainforest bird. Perspect. Ecol. Conserv. 2017, 15, 3–9. [Google Scholar] [CrossRef]
- Avery, M.L. Rusty blackbird (Euphagus carolinus), Version 1.0. In Birds of the World; Poole, A.F., Ed.; Cornell Lab of Ornithology: Ithaca, NY, USA, 2020. [Google Scholar]
- Stralberg, D.; Matsuoka, S.M.; Handel, C.M.; Bayne, E.M.; Schmiegelow, F.K.A.; Hamman, A. Biogeography of boreal passerine range dynamics in western North America: Past, present, and future. Ecography 2017, 40, 1050–1066. [Google Scholar] [CrossRef]
- DaCosta, J.M.; Sorenson, M.D. Amplification biases and consistent recovery of loci in a double-digest RAD-seq protocol. PLoS ONE 2014, 9, e106713. [Google Scholar] [CrossRef] [PubMed]
- Sonsthagen, S.A.; Wilson, R.E.; Matsuoka, S.M.; Johnson, J.A.; Demarest, D.W.; Stralberg, D.; Powell, L.L. Rusty blackbird (Euphagus carolinus) genetic data, North America. U.S. Geological Survey Data Release. 2021. [Google Scholar] [CrossRef]
- BU-RAD-seq ddRAD-seq Pipeline. Available online: http://github.com/BU-RAD-seq/ddRAD-seq-Pipeline (accessed on 30 June 2020).
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, W.E.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 2015, 403–410. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavretsky, P.; DaCosta, J.M.; Hernández-Baños, B.E.; Engils, A., Jr.; Sorenson, M.D.; Peters, J.L. Speciation genomics and a role for the Z chromosome in the early stages of divergence between Mexican ducks and mallards. Mol. Ecol. 2015, 25, 661–674. [Google Scholar] [CrossRef]
- Dufort, M.J.; Barker, F.K. Range dynamics, rather than convergent selection, explain the mosaic distribution of red-winged blackbird phenotypes. Ecol. Evol. 2013, 3, 4910–4924. [Google Scholar] [CrossRef]
- Sonsthagen, S.A.; Talbot, S.L.; McCracken, K.G. Genetic characterization of common eiders (Somateria mollissima) breeding on the Yukon-Kuskokwim Delta, Alaska. Condor 2007, 109, 879–894. [Google Scholar] [CrossRef]
- BU-RAD-Seq Out-Conversions. Available online: http://github.com/BU-RAD-seq/Out-Conversions (accessed on 30 June 2020).
- Schneider, S.; Roessli, D.; Excoffier, L. ARLEQUIN Version 2.0: A Software for Population Genetic Analyses; Genetics and Biometry Laboratory, University of Geneva: Geneva, Switzerland, 2000. [Google Scholar]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selections. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Tajima, F. The effect of change in population size on DNA polymorphism. Genetics 1989, 123, 597–601. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Sykes, B.C.; Richards, M.B. Mitochondrial portraits of human populations. Genetics 1995, 152, 743–753. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of the mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Petkova, D.; Novembre, J.; Stephens, M. Visualizing spatial population structure with estimated effective migration surfaces. Nat. Genet. 2016, 48, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Dray, S.; Dufour, A.-B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T. adegent: An R package for the multivariate analysis of genetic markers. Bioinfomatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinfomatics 2011, 12, 246. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Admixture 1.22 Software Manual; Cold Spring Harbor Lab: New York, NY, USA, 2012. [Google Scholar]
- Zhou, H.; Alexander, D.; Lange, K. A quasi-Newton acceleration for high-dimensional optimization algorithms. Stat. Comput. 2011, 21, 261–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Francis, R.M. Pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Malinksy, M.; Trucchi, E.; Lawson, D.J.; Falush, D. RADpainter and fineRADstructure: Population inference from RADseq data. Mol. Biol. Evol. 2018, 35, 1284–1290. [Google Scholar]
- fineRADstructure. Available online: http://cichlid.gurdon.cam.ac.uk/fineRADstructure.html (accessed on 30 June 2020).
- EEMS. Available online: https://github.com/dipetkov/eems (accessed on 30 June 2020).
- Google Maps API v3 Tool. Available online: http://www.birdtheme.org/useful/v3tool.html (accessed on 30 June 2020).
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef]
- Waples, R.S.; Do, C. LDNE: A program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Resour. 2008, 8, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Luikart, G.; Ryman, N.; Tallmon, D.A.; Schwartz, M.K.; Allendorf, F.W. Estimation of census and effective population sizes: The increasing usefulness of DNA-based approaches. Conserv. Genet. 2010, 11, 355–373. [Google Scholar] [CrossRef]
- Nomura, T. Estimation of effective number breeders from molecular coancestry of single cohort sample. Evol. Appl. 2008, 1, 462–474. [Google Scholar] [CrossRef]
- Waples, R.S.; England, P.R. Estimating contemporary effective population size on the basis of linkage disequilibrium in the face of migration. Genetics 2011, 189, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Elith, J.; Leathwick, J.R.; Hastie, T. A working guide to boosted regression trees. J. Anim. Ecol. 2008, 77, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Stralberg, D.; Matsuoka, S.M.; Hamann, A.; Bayne, E.M.; Sólymos, P.; Schmiegelow, F.K.A.; Wang, X.; Cumming, S.G.; Song, S.J. Projecting boreal bird responses to climate change: The signal exceeds the noise. Ecol. Appl. 2015, 25, 52–69. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Hamann, A.; Spittlehouse, D.L.; Murdock, T.Q. ClimateWNA—high-resolution spatial climate data for western North America. J. Appl. Meteorol. Climatol. 2012, 51, 16–29. [Google Scholar] [CrossRef] [Green Version]
- Hogg, E.H. Temporal scaling of moisture and the forest-grassland boundary in western Canada. Agric. For. Meteorol. 1997, 84, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.R.; Hamann, A. Predicting potential climate change impacts with bioclimate envelope models: A palaeoecological perspective. Glob. Ecol. Biogeogr. 2012, 21, 121–133. [Google Scholar] [CrossRef]
- Waples, R.S.; Do, C. Linage disequilibrium estimates of contemporary Ne using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evol. Appl. 2010, 3, 244–262. [Google Scholar] [CrossRef]
- CEC. Ecological Regions of North America: Toward a Common Perspective; Commission for Environmental Cooperation: Montreal, QC, Canada, 1997; 60p. [Google Scholar]
- Ehlers, J.; Gibbard, P.L.; Hughes, P.D. (Eds.) Quaternary Glaciations—Extent and Chronology; Elsevier: Amsterdam, The Netherlands, 2011; 1108p. [Google Scholar]
- LGM Glaciation Extends. Available online: https://crc806db.uni-koeln.de/layer/show/6 (accessed on 18 February 2021).
- Dyke, A.S. An outline of North American deglaciation with emphasis on central and northern Canada. In Quaternary Glaciations—Extent and Chronology, Part II: North America, Developments in Quaternary Science; Ehlers, J., Gibbard, P.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 2b, pp. 373–424. [Google Scholar]
- Johnson, N.K.; Cicero, C. New mitochondrial DNA data affirm the importance of Pleistocene speciation in North American birds. Evolution 2004, 58, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Weir, J.; Schluter, D. Ice sheets promote speciation in boreal birds. Proc. R. Soc. Lond. B Biol. Sci. 2004, 271, 1881–1887. [Google Scholar] [CrossRef] [Green Version]
- Boulet, M.; Gibbs, H.L.; Hobson, K.A. Integrated analysis of genetic, stable isotope, and banding data reveal migratory connectivity and flyways in the northern yellow warbler (Dendroica petechia; Aestiva group). Ornithol. Monogr. 2006, 61, 29–78. [Google Scholar] [CrossRef]
- Bay, R.A.; Harrigan, R.J.; Underwood, V.L.; Gibbs, H.L.; Smith, T.B.; Ruegg, K. Genomic signals of selection predict climate-driving population declines in a migratory bird. Science 2018, 359, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.E.; Irwin, J.H.; Smith, T.B. Genetic variation and seasonal migratory connectivity in Wilson’s warblers (Wilsonia pusilla): Species-level differences in nuclear DNA between western and eastern populations. Mol. Ecol. 2011, 20, 3102–3115. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.A.; Carpenter, A.M.; Friesen, V.L.; Burg, T.M. A comparison of neutral genetic differentiation and genetic diversity among migratory and resident populations of golden-crowned kinglets (Regulus satrapa). J. Ornithol. 2020, 161, 509–519. [Google Scholar] [CrossRef]
- Gérardi, S.; Jaramillo-Correa, J.P.; Beaulieu, J.; Bousquet, J. From glacial refugia to modern populations: New assemblages of organelle genomes generated by differential cytoplasmic gene flow in transcontinental black spruce. Mol. Ecol. 2010, 19, 5265–5280. [Google Scholar] [CrossRef]
- Matsuoka, S.M.; Shaw, D.; Sinclair, P.H.; Johnson, J.A.; Corcoran, R.M.; Dau, N.C.; Meyers, P.M.; Rojek, N.A. Nesting ecology of Rusty Blackbirds in Alaska and Canada. Condor 2010, 112, 810–824. [Google Scholar] [CrossRef]
- Jaramillo-Correa, J.P.; Beaulieu, J.; Bousquet, J. Variation in mitochondrial DNA reveals multiple distant glacial refugia in black spruce (Picea mariana), a transcontinental North American conifer. Mol. Ecol. 2004, 13, 2735–2747. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.R.; Hamann, A. Glacial refugia and modern genetic diversity of 22 western North American tree species. Proc. R. Soc. B 2015, 282, 2014–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, G.M. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 1996, 58, 247–276. [Google Scholar] [CrossRef]
- Macfarlane, C.B.A.; Natola, L.; Brown, M.W.; Burg, T.M. Population genetic isolation and limited connectivity in the purple finch (Haemorphous purpureus). Ecol. Evol. 2016, 6, 8304–8317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FitzGerald, A.M.; Whitaker, D.M.; Ralston, J.; Kirchman, J.J.; Warkentin, I.G. Taxonomy and distribution of the imperilled Newfoundland gray-cheeked thrush, Catharus minimus minimus. Avian Conserv. Ecol. 2017, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Hindley, J.; Graham, B.A.; Burg, T.M. Pleistocene glacial cycles and physical barriers influence phylogeographic structure in black-capped chickadees (Poecile atricapillus), a widespread North American passerine. Can. J. Zool. 2018, 96, 1366–1377. [Google Scholar] [CrossRef] [Green Version]
- Friis, G.; Aleixandre, P.; Rodríguez-Estrella, R.; Navarro-Sigüenza, A.G.; Milá, B. Rapid postglacial diversification and long-term stasis within the songbird genus Junco: Phylogeographic and phylogenomic evidence. Mol. Ecol. 2016, 25, 6175–6195. [Google Scholar] [CrossRef] [Green Version]
- Ruegg, K. Genetic, morphological, and ecological characterization of a hybrid zone that spans a migratory divide. Evolution 2008, 62, 452–466. [Google Scholar] [CrossRef]
- Delmore, K.E.; Fox, J.W.; Irwin, D.E. Dramatic intraspecific differences in migratory routes, stopover sites and wintering areas, revealed using light-level geolocators. Proc. R. Soc. B 2012, 279, 4582–4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmore, K.E.; Irwin, D.E. Hybrid songbirds employ intermediate routes in a migratory divide. Ecol. Lett. 2014, 17, 1211–1218. [Google Scholar] [CrossRef]
- Cohen, E.B.; Rushing, C.R.; Moore, F.R.; Hallworth, M.T.; Hostetler, J.A.; Ramirez, M.G.; Marra, P.P. The strength of migratory connectivity for birds en route to breeding through the Gulf of Mexico. Ecography 2019, 42, 658–669. [Google Scholar] [CrossRef] [Green Version]
- Machtans, C.S.; Van Wilgenburg, S.L.; Armer, L.A.; Hobson, K.A. Retrospective comparison of the occurrence and abundance of rusty blackbird in the Mackenzie Valley, Northwest Territories. Avian Conserv. Ecol. 2007, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Belisle, M.; Desrochers, A.; Fortin, M.J. Influence of forest cover on the movements of forest birds: A homing experiment. Ecology 2001, 82, 1893–1904. [Google Scholar]
- Matthysen, E.; Adriaensen, F.; Dhondt, A.A. Dispersal of nuthatches, Sitta europaea, in a highly fragmented forest habitat. Oikos 1995, 72, 375–381. [Google Scholar] [CrossRef]
- Matthysen, E.; Currie, D. Habitat fragmentation reduces disperser success in juvenile Sitta europaea: Evidence from patterns of territory establishment. Ecography 1996, 19, 67–72. [Google Scholar] [CrossRef]
- Stralberg, D.; Berteaux, D.; Drever, C.R.; Drever, M.; Naujokaitis-Lewis, I.; Schmiegelow, F.K.A.; Tremblay, J.A. Conservation planning for boreal birds in a changing climate: A framework for action. Avian Conserv. Ecol. 2019, 14, 13. [Google Scholar] [CrossRef]
- Rehfeldt, G.E.; Crookston, N.L.; Sáenz-Romero, C.; Campbell, E.M. North American vegetation model for land-use planning in a changing climate: A solution to large classification problems. Ecol. Appl. 2012, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Bateman, B.L.; Taylor, L.; Wilsey, C.; Wu, J.; LeBaron, G.S.; Langham, G. Risk to North American birds from climate change-related threats. Conserv. Sci. Pract. 2020, 2, e243. [Google Scholar] [CrossRef]
- Bateman, B.L.; Wilsey, C.; Taylor, L.; Wu, J.; LeBaron, G.S.; Langham, G. North American birds require mitigation and adaptation to reduce vulnerability to climate change. Conserv. Sci. Pract. 2020, 2, e242. [Google Scholar] [CrossRef]
- Fitzpatrick, M.C.; Keller, S.R. Ecological genomics meets community-level modelling of biodiversity: Mapping the genomic landscape of current and future environmental adaptation. Ecol. Lett. 2015, 18, 1–16. [Google Scholar] [CrossRef]
- Greenberg, R.S. Bye bye blackbird: Rusty blackbirds are vanishing from our southern swamps and northern forests. Zoogoer 2008, 37, 11–15. [Google Scholar]
- Rushing, C.S.; Ryder, T.B.; Marra, P.P. Quantifying drivers of population dynamics for a migratory bird throughout the annual cycle. Proc. R. Soc. B 2016, 283, 20152846. [Google Scholar] [CrossRef]
- Rushing, C.S.; Hostetler, J.A.; Sillett, T.S.; Marra, P.P.; Rotenberg, J.A.; Ryder, T.B. Spatial and temporal drivers of avian population dynamics across the annual cycle. Ecology 2017, 98, 2837–2850. [Google Scholar] [CrossRef]
- Williams, C.K.; Castelli, P.M. 2012. A historical perspective of the connectivity between waterfowl research and management. In Wildlife Science: Connecting Research with Management; Sands, J.P., Brennan, L.A., DeMaso, S.J., Schnupp, M.J., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 155–178. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | ddRAD | mtDNA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| π − A | π − Z | Ne | n | H | π | h | Fs | D | n | ||
| 1 | Bethel, Alaska, USA | 0.0065 | 0.0041 | – | 6 | 4 | 0.0046 (0.0033) | 0.867 (0.129) | 0.3 | −0.4 | 6 |
| 2 | Anchorage, Alaska, USA | 0.0069 | 0.0048 | 46.7 (38.5–55.7) | 24 | 13 | 0.0050 (0.0031) | 0.899 (0.046) | −5.1* | −1.2 | 24 |
| 3 | Cordova, Alaska, USA | 0.0065 | 0.0045 | – | 6 | 2 | 0.0012 (0.0013) | 0.333 (0.215) | 1.6 | −1.1 | 6 |
| 4 | Tanana, Alaska, USA | 0.0067 | 0.0048 | – | 9 | 4 | 0.0022 (0.0017) | 0.694 (0.147) | 0.0 | −0.8 | 9 |
| 5 | Tetlin NWR, Alaska, USA | 0.0070 | 0.0049 | 48.7 (36.6–62.5) | 30 | 14 | 0.0038 (0.0025) | 0.893 (0.040) | −6.3* | −1.4 | 31 |
| 6 | Yukon Flats, Alaska, USA | 0.0070 | 0.0049 | 21.3 (18.4–24.5) | 25 | 15 | 0.0048 (0.0030) | 0.952 (0.029) | −9.2* | −0.8 | 22 |
| 7 | Yukon Territory, Canada | 0.0058 | 0.0043 | – | 4 | 3 | 0.0034 (0.0029) | 0.833 (0.222) | 0.0 | 1.1 | 4 |
| 8 | Alberta, Canada | 0.0069 | 0.0048 | 26.5 (23.5–29.7) | 21 | 12 | 0.0039 (0.0025) | 0.922 (0.035) | −5.8* | −1.1 | 22 |
| 9 | Manitoba, Canada | 0.0066 | 0.0048 | – | 7 | 7 | 0.0063 (0.0042) | 1.000 (0.076) | −3.6* | −0.4 | 7 |
| 10 | Ontario, Canada | 0.0070 | 0.0048 | 41.3 (33.9–49.3) | 23 | 15 | 0.0036 (0.0024) | 0.949 (0.028) | −11.6* | −1.2 | 23 |
| 11 | Newfoundland, Canada | 0.0062 | 0.0037 | 19.1 (16.7–21.6) | 10 | 4 | 0.0040 (0.0027) | 0.691 (0.128) | 0.9 | −0.4 | 11 |
| 12 | Nova Scotia/New Brunswick, Canada | 0.0061 | 0.0040 | – | 5 | 7 | 0.0069 (0.0045) | 1.000 (0.076) | −3.8* | 0.1 | 7 |
| 13 | New Hampshire/Vermont, USA | 0.0068 | 0.0046 | 22.4 (19.4–25.6) | 28 | 13 | 0.0047 (0.0029) | 0.878 (0.041) | −5.2* | −0.3 | 28 |
| All populations | 0.0061 | 0.0050 | – | 198 | – | – | – | – | – | 200 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, R.E.; Matsuoka, S.M.; Powell, L.L.; Johnson, J.A.; Demarest, D.W.; Stralberg, D.; Sonsthagen, S.A. Implications of Historical and Contemporary Processes on Genetic Differentiation of a Declining Boreal Songbird: The Rusty Blackbird. Diversity 2021, 13, 103. https://doi.org/10.3390/d13030103
Wilson RE, Matsuoka SM, Powell LL, Johnson JA, Demarest DW, Stralberg D, Sonsthagen SA. Implications of Historical and Contemporary Processes on Genetic Differentiation of a Declining Boreal Songbird: The Rusty Blackbird. Diversity. 2021; 13(3):103. https://doi.org/10.3390/d13030103
Chicago/Turabian StyleWilson, Robert E., Steven M. Matsuoka, Luke L. Powell, James A. Johnson, Dean W. Demarest, Diana Stralberg, and Sarah A. Sonsthagen. 2021. "Implications of Historical and Contemporary Processes on Genetic Differentiation of a Declining Boreal Songbird: The Rusty Blackbird" Diversity 13, no. 3: 103. https://doi.org/10.3390/d13030103
APA StyleWilson, R. E., Matsuoka, S. M., Powell, L. L., Johnson, J. A., Demarest, D. W., Stralberg, D., & Sonsthagen, S. A. (2021). Implications of Historical and Contemporary Processes on Genetic Differentiation of a Declining Boreal Songbird: The Rusty Blackbird. Diversity, 13(3), 103. https://doi.org/10.3390/d13030103