
Genetic Consequences of Fence Confinement in a Population of White-Tailed Deer

 ,
,  ,
,
Abstract
:1. Introduction
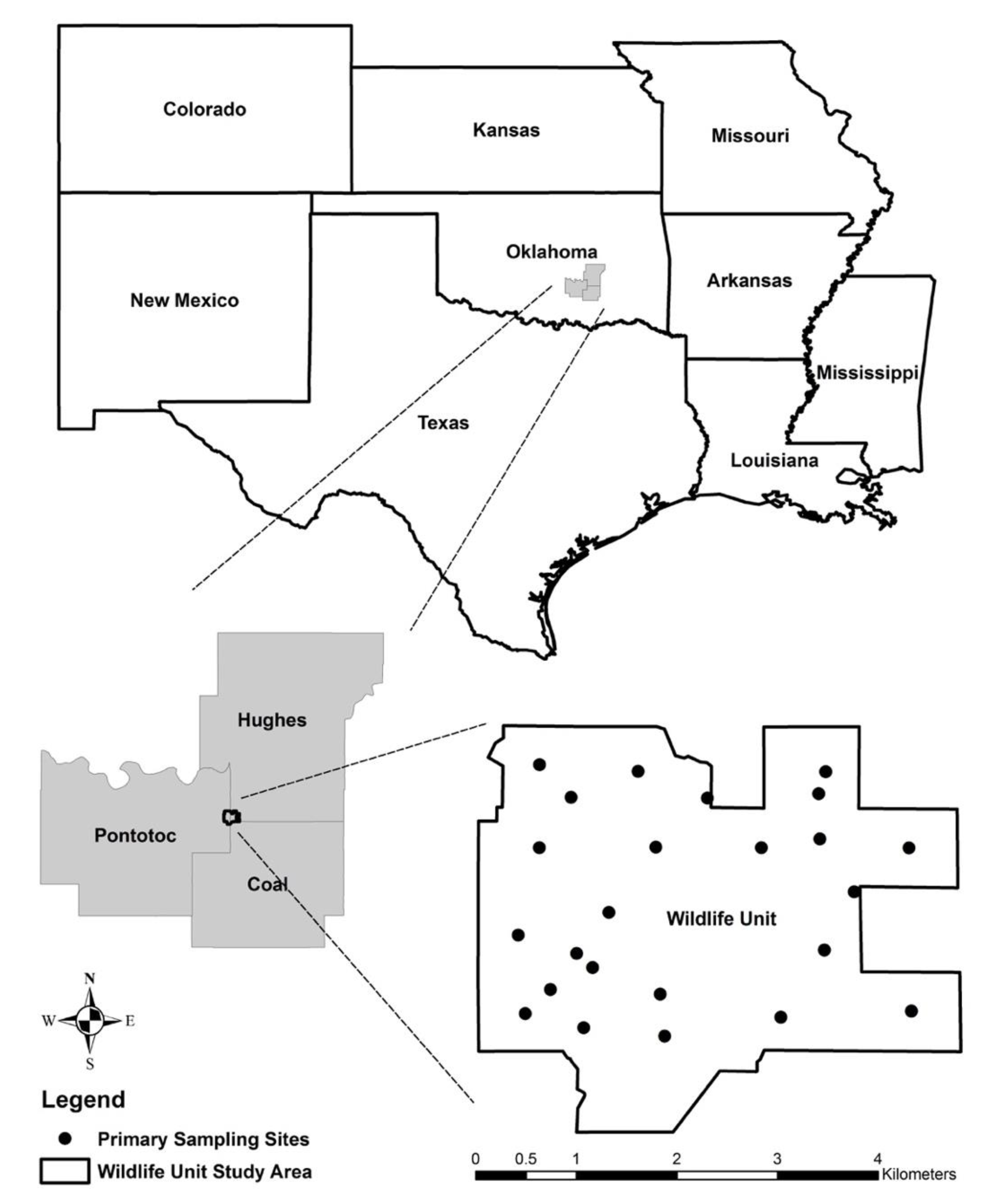
2. Materials and Methods
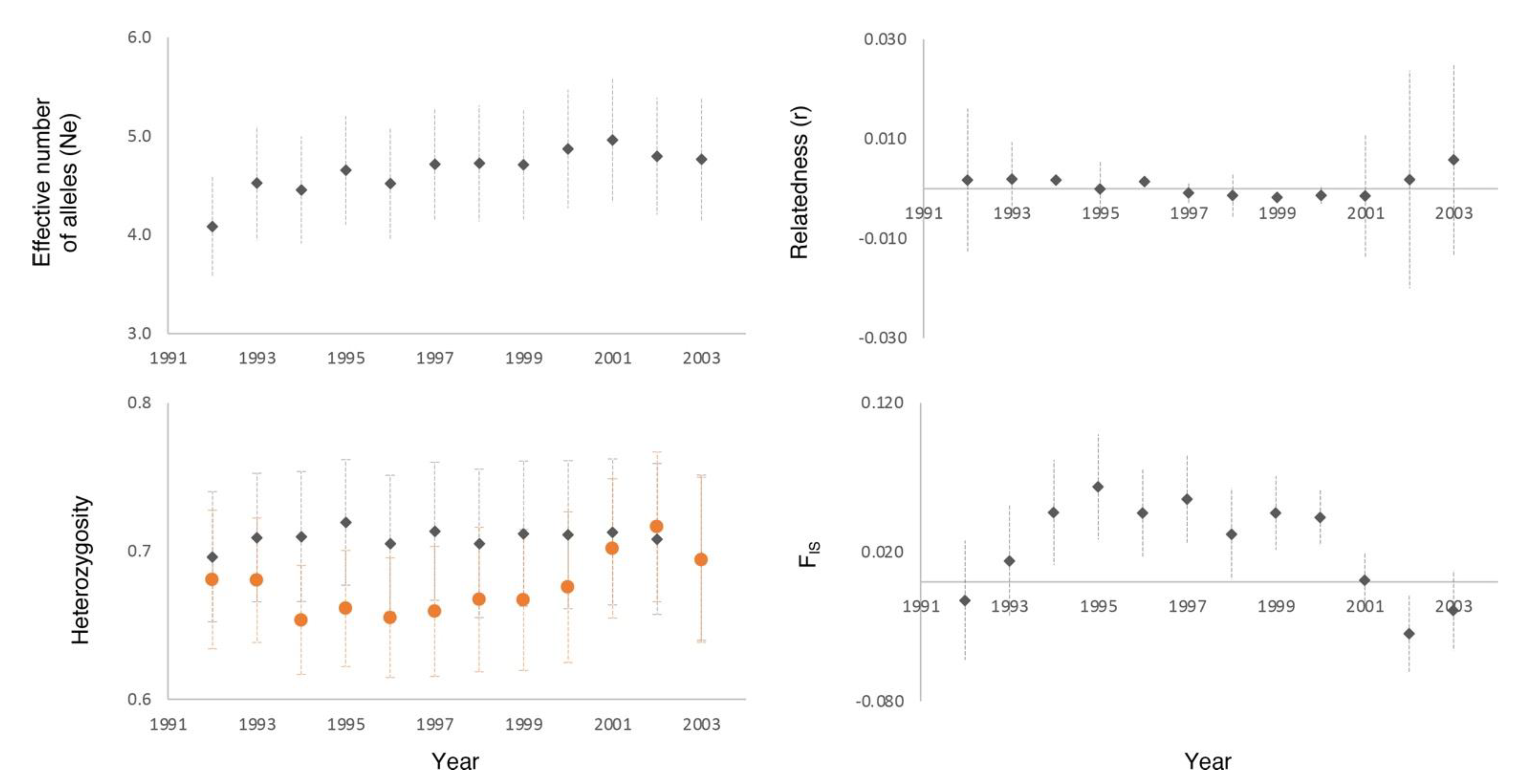
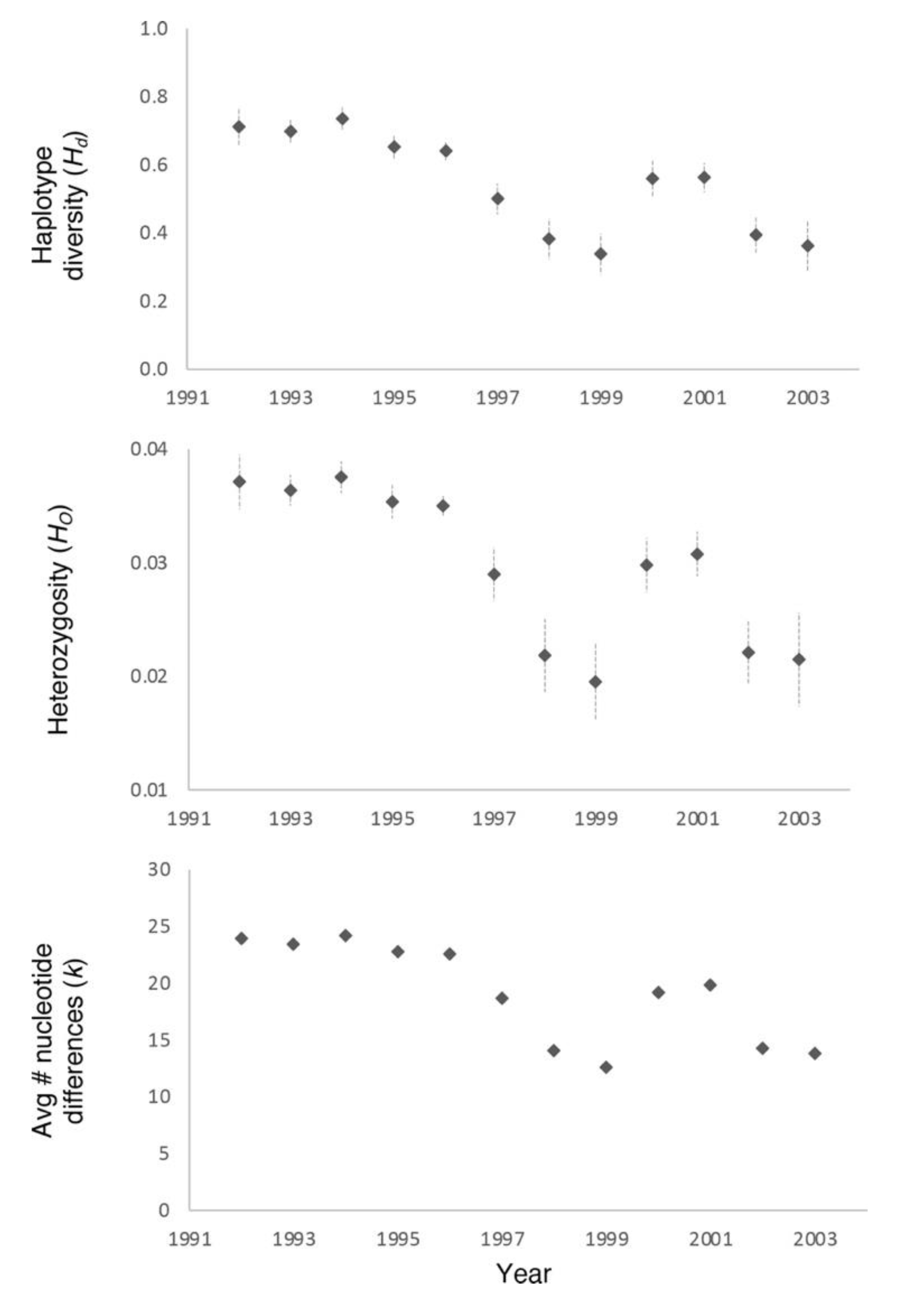
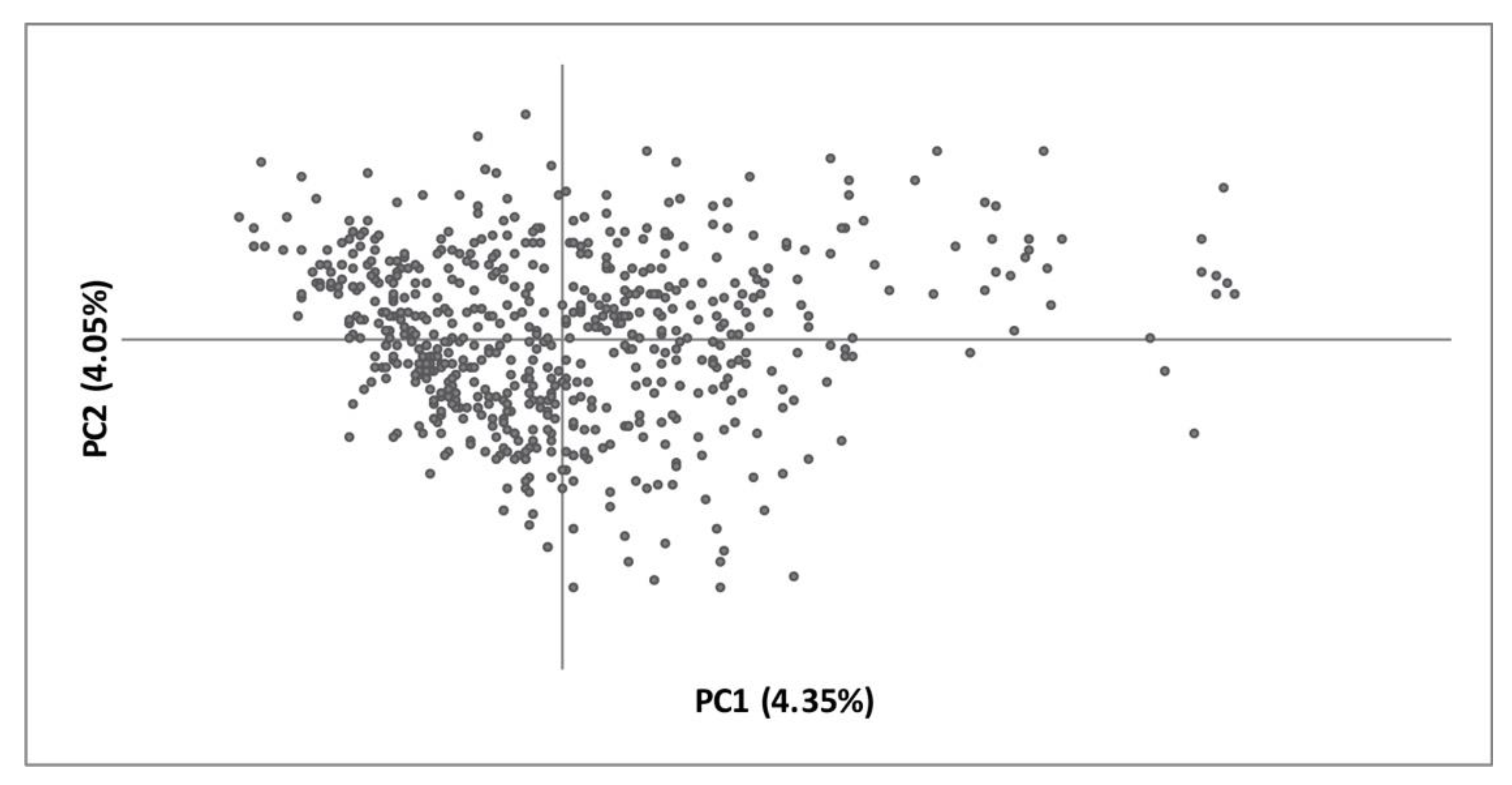
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, R.; Williams, E. Fences and Between Fences: Cultural, Historical, and Smithsonian Perspectives. J. Southwest 2008, 50, 243–261. [Google Scholar] [CrossRef]
- Morris, L.R.; Rowe, R.J. Historical land use and altered habitats in the Great Basin. J. Mammal. 2014, 95, 1144–1156. [Google Scholar] [CrossRef] [Green Version]
- Jakes, A.F.; Jones, P.F.; Paige, L.C.; Seidler, R.G.; Huijser, M.P. A fence runs through it: A call for greater attention to the in-fluence of fences on wildlife and ecosystems. Biol. Conserv. 2018, 227, 310–318. [Google Scholar] [CrossRef]
- McInturff, A.; Xu, W.; Wilkinson, C.E.; Dejid, N.; Brashares, J.S. Fence Ecology: Frameworks for Understanding the Ecological Effects of Fences. Bioscience 2020, 70, 971–985. [Google Scholar] [CrossRef]
- Clevenger, A.P.; Chruszca, B.; Gunson, K.E. Highway mitigation fencing reduces wildlife-vehicle collisions. Wildl. Soc. Bull. 2001, 29, 646–653. [Google Scholar]
- Moseby, K.; Read, J. The efficacy of feral cat, fox and rabbit exclusion fence designs for threatened species protection. Biol. Conserv. 2006, 127, 429–437. [Google Scholar] [CrossRef]
- Mysterud, A.; Rolandsen, C.M. Fencing for wildlife disease control. J. Appl. Ecol. 2019, 56, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Cornwall, W. To save caribou, Alberta wants to fence them in: Controversial proposal envisions the construction of a massive, predator-free pen. Science 2016, 353, 333. [Google Scholar] [CrossRef] [Green Version]
- Ezenwa, V.O. Parasite infection rates of impala (Aepyceros melampus) in fenced game reserves in relation to reserve charac-teristics. Biol. Conserv. 2004, 118, 397–401. [Google Scholar] [CrossRef]
- Ferguson, K.; Hanks, J. Fencing Impacts: A Review of the Environmental, Social, and Economic Impacts of Game and Veterinary Fencing in Africa with Particular Reference to the Great Limpopo and Kavango Zambezi Transfrontier Conservation Areas; University of Pretoria, Mammal Research Institute: Pretoria, Africa, 2010. [Google Scholar]
- Ferronato, B.O.; Roe, J.H.; Georges, A. Reptile bycatch in a pest-exclusion fence established for wildlife reintroductions. J. Nat. Conserv. 2014, 22, 577–585. [Google Scholar] [CrossRef]
- Pekor, A.; Miller, J.R.; Flyman, M.V.; Kasiki, S.; Kesch, M.K.; Miller, S.M.; Uiseb, K.; van der Merve, V.; Lindsey, P.A. Fencing Africa’s protected areas: Costs, benefits, and management issues. Biol. Conserv. 2019, 229, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Laskin, D.N.; Watt, D.; Whittington, J.; Heuer, K. Designing a fence that enables free passage of wildlife while containing reintroduced bison: A multispecies evaluation. Wildl. Biol. 2020, 2020, 751. [Google Scholar] [CrossRef]
- Vercauteren, K.C.; Lavelle, M.J.; Hygnstrom, S.E. Fences and Deer-Damage Management: A Review of Designs and Efficacy. Wildl. Soc. Bull. 2006, 34, 191–200. [Google Scholar] [CrossRef]
- Nielsen, C.K.; Nelson, S.J.; Porter, W.F. Emigration of deer from a partial enclosure. Wildl. Soc. Bull. 1997, 25, 282–290. [Google Scholar]
- Webb, S.L.; Gee, K.L.; DeMarais, S.; Strickland, B.K.; Deyoung, R.W. Efficacy of a 15-Strand High-Tensile Electric Fence to Control White-tailed Deer Movements. Wildl. Biol. Pract. 2009, 5, 45–57. [Google Scholar] [CrossRef]
- Webb, S.L.; Gee, K.L.; Wang, G. Survival and fidelity of an enclosed white-tailed deer population using capture–recapture-reporting data. Popul. Ecol. 2009, 52, 81–88. [Google Scholar] [CrossRef]
- Webb, S.L.; Gee, K.L. Annual survival and site fidelity of free-ranging white-tailed deer (Odocoileus virginianus): Comparative demography before (1983–1992) and after (1993–2005) spatial confinement. Integr. Zool. 2014, 9, 24–33. [Google Scholar] [CrossRef]
- Ozoga, J.J.; Verme, L.J. Physical and Reproductive Characteristics of a Supplementally-Fed White-Tailed Deer Herd. J. Wildl. Manag. 1982, 46, 281. [Google Scholar] [CrossRef]
- Demarais, S.; DeYoung, R.W.; Lyon, L.J.; Williams, E.S.; Williamson, S.J.; Wolfe, G.J. Biological and social issues related to confinement of wild ungulates. Wildl. Soc. Tech. Rev. 2002, 2–3, 1–29. [Google Scholar]
- Connolly, T.A.; Day, T.D.; King, C.M. Estimating the potential for reinvasion by mammalian pests through pest-exclusion fencing. Wildl. Res. 2009, 36, 410–421. [Google Scholar] [CrossRef]
- Vanak, A.T.; Thaker, M.; Slotow, R. Do fences create an edge-effect on the movement patterns of a highly mobile mega-herbivore? Biol. Conserv. 2010, 143, 2631–2637. [Google Scholar] [CrossRef]
- Wright, S. Evolution in Mendelian Populations. Genetics 1931, 16, 97–159. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.F.; Waller, D.M. Inbreeding effects in wild populations. Trends Ecol. Evol. 2002, 17, 230–241. [Google Scholar] [CrossRef]
- Falconer, D.S. Introduction to Quantitative Genetics, 2nd ed.; Longman Inc.: New York, NY, USA, 1981. [Google Scholar]
- Deyoung, R.W.; DeMarais, S.; Honeycutt, R.L.; Gee, K.L.; Gonzales, R.A. Social Dominance and Male Breeding Success in Captive White-Tailed Deer. Wildl. Soc. Bull. 2006, 34, 131–136. [Google Scholar] [CrossRef]
- Gee, K.L.; Porter, M.D.; Demarais, S.; Bryant, F.C.; Van Vreede, G. White-Tailed Deer: Their Foods and Management in the Cross Timbers, 2nd ed.; Samuel Roberts Noble Foundation: Ardmore, OK, USA, 1994. [Google Scholar]
- Webb, S.L.; Gee, K.L.; Deyoung, R.W.; Harju, S.M. Variance component analysis of body mass in a wild population of deer (Odocoileus virginianus): Results from two decades of research. Wildl. Res. 2013, 40, 588–598. [Google Scholar] [CrossRef] [Green Version]
- DeYoung, R.W. Effects of Social and Population Characteristics on the Reproductive Success of Male White-Tailed Deer. Ph.D. Thesis, Mississippi State University, Starkville, MI, USA, 2004. [Google Scholar]
- Anderson, J.D.; Honeycutt, R.L.; Gonzales, R.A.; Gee, K.L.; Skow, L.C.; Gallagher, R.L.; Honeycutt, D.A.; Deyoung, R.W. Development of Microsatellite DNA Markers for the Automated Genetic Characterization of White-Tailed Deer Populations. J. Wildl. Manag. 2002, 66, 67. [Google Scholar] [CrossRef]
- DeYoung, R.W.; Demarais, S.; Honeycutt, R.L.; Gonzales, R.A.; Gee, K.L.; Anderson, J.D. Evaluation of a DNA microsatellite panel useful for genetic exclusion studies in white-tailed deer. Wildl. Soc. Bull. 2003, 31, 220–232. [Google Scholar]
- Lischer, H.E.L.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2011, 28, 298–299. [Google Scholar] [CrossRef] [Green Version]
- Bickham, J.W.; Patton, J.C.; Loughlin, T.R. High Variability for Control-Region Sequences in a Marine Mammal: Implications for Conservation and Biogeography of Steller Sea Lions (Eumetopias jubatus). J. Mammal. 1996, 77, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 24, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Royal Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Rozas, J.; Mata, F.A.; Del Barrio, S.J.C.; Rico, G.S.; Librado, P.; Onsins, R.S.E.; Gracia, S.A. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Corander, J.; Waldmann, P.; Sillanpää, M.J. Bayesian analysis of genetic differentiation between populations. Genetics 2003, 163, 367–374. [Google Scholar]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Loiselle, B.A.; Sork, V.L.; Nason, J.; Graham, C. Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am. J. Bot. 1995, 82, 1420–1425. [Google Scholar] [CrossRef]
- Lynch, M.; Ritland, K. Estimation of pairwise relatedness with molecular markers. Genetetics 1999, 152, 1753–1766. [Google Scholar]
- Miller, W.L.; Edson, J.; Pietrandrea, P.; Butterworth, M.C.; Walter, W.D. Identification and evaluation of a core microsatellite panel for use in white-tailed deer (Odocoileus virginianus). BMC Genet. 2019, 20, 49. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.K.; McKelvey, K.S. Why sampling scheme matters: The effect of sampling scheme on landscape genetic results. Conserv. Genet. 2009, 10, 441–452. [Google Scholar] [CrossRef]
- Latch, E.K.; Dharmarajan, G.; Glaubitz, J.C.; Rhodes, O.E. Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conserv. Genet. 2006, 7, 295–302. [Google Scholar] [CrossRef]
- Sutherland, W.J.; Barnard, P.; Broad, S.; Clout, M.; Connor, B.; Côté, I.M.; Dicks, L.V.; Doran, H.; Entwistle, A.C.; Fleishman, E.; et al. A 2017 Horizon Scan of Emerging Issues for Global Conservation and Biological Diversity. Trends Ecol. Evol. 2017, 32, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.F.; De Young, R.W.; Campbell, T.A.; Laseter, B.R.; Ford, W.M.; Miller, K.V. Fine-scale genetic and social structuring in a central Appalachian white-tailed deer herd. J. Mammal. 2010, 91, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Cullingham, C.I.; Merrill, E.H.; Pybus, M.J.; Bollinger, T.K.; Wilson, G.A.; Coltman, D.W. Broad and fine-scale genetic analysis of white-tailed deer populations: Estimating the relative risk of chronic wasting disease spread. Evol. Appl. 2010, 4, 116–131. [Google Scholar] [CrossRef]
- De La Reyna, R.X.F.; Lobato, C.R.D.; Bracamonte, P.G.M.; Rincón, S.A.M.; Deyoung, R.W.; León, F.J.G.D.; Vera, A.W. Genetic diversity and structure among subspecies of white-tailed deer in Mexico. J. Mammal. 2012, 93, 1158–1168. [Google Scholar] [CrossRef] [Green Version]
- Kekkonen, J.; Wikström, M.; Brommer, J.E. Heterozygosity in an isolated population of a large mammal founded by four indi-viduals is predicted by an individual-based genetic model. PLoS ONE 2012, 7, 43482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanova, V.L.; Hughes, P.T.; Hoffman, E.A. Combining genetic structure and demographic analyses to estimate persistence in endangered Key deer (Odocoileus virginianus clavium). Conserv. Genet. 2017, 18, 1061–1076. [Google Scholar] [CrossRef]
- Hopken, M.W.; Lum, T.M.; Meyers, P.M.; Piaggio, A.J. Molecular assessment of translocation and management of an endan-gered subspecies of white-tailed deer (Odocoileus virginianus). Conserv. Genet. 2015, 16, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Blanchong, J.A.; Sorin, A.B.; Scribner, K.T. Genetic diversity and population structure in urban white-tailed deer. J. Wildl. Manag. 2013, 77, 855–862. [Google Scholar] [CrossRef]
- Webb, S.L.; DeYoung, R.W.; Demarais, S.; Strickland, B.K.; Gee, K.L. Testing a local inbreeding hypothesis as a cause of ob-served antler characteristics in managed populations of white-tailed deer. Diversity 2021, 13, 116. [Google Scholar] [CrossRef]
- Deyoung, R.W.; DeMarais, S.; Gee, K.L.; Honeycutt, R.L.; Hellickson, M.W.; Gonzales, R.A. Molecular Evaluation of the White-tailed Deer (Odocoileus Virginianus) Mating System. J. Mammal. 2009, 90, 946–953. [Google Scholar] [CrossRef] [Green Version]
- Hayward, M.W.; Kerley, G.I.H. Fencing for conservation: Restriction of evolutionary potential or a riposte to threatening processes? Biol. Conserv. 2009, 142, 1–13. [Google Scholar] [CrossRef]
- Vercauteren, K.C.; Lavelle, M.J.; Seward, N.W.; Fischer, J.W.; Phillips, G.E. Fence-Line Contact Between Wild and Farmed White-Tailed Deer in Michigan: Potential for Disease Transmission. J. Wildl. Manag. 2007, 71, 1603–1606. [Google Scholar] [CrossRef] [Green Version]
- Vercauteren, K.C.; Lavelle, M.J.; Seward, N.W.; Fischer, J.W.; Phillips, G.E. Fence-line contact between wild and farmed white-tailed deer in Colorado: Potential for disease transmission. J. Wildl. Manag. 2007, 71, 1594–1602. [Google Scholar] [CrossRef]
- Grear, D.A.; Samuel, M.D.; Scribner, K.T.; Weckworth, B.V.; Langenberg, J.A. Influence of genetic relatedness and spatial proximity on chronic wasting disease infection among female white-tailed deer. J. Appl. Ecol. 2010, 47, 532–540. [Google Scholar] [CrossRef]
- Magle, S.B.; Samuel, M.D.; Van Deelen, T.R.; Robinson, S.J.; Mathews, N.E. Evaluating Spatial Overlap and Relatedness of White-tailed Deer in a Chronic Wasting Disease Management Zone. PLoS ONE 2013, 8, e56568. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | AR | HO | HE | FIS | Pval(FIS ≠ 0) |
|---|---|---|---|---|---|
| BovPRL | 2.00 | 0.309 | 0.294 | −0.053 | 0.187 |
| Cervid1 | 14.81 | 0.868 | 0.867 | −0.002 | 0.896 |
| ILSTS011 | 9.98 | 0.775 | 0.844 | 0.082 | 0 |
| INRA011 | 5.99 | 0.676 | 0.667 | −0.013 | 0.581 |
| N | 19.17 | 0.763 | 0.871 | 0.124 | 0 |
| Q | 19.38 | 0.810 | 0.841 | 0.037 | 0.020 |
| BL25 | 4.00 | 0.498 | 0.530 | 0.059 | 0.053 |
| BM6438 | 12.79 | 0.657 | 0.817 | 0.195 | 0 |
| BM848 | 12.82 | 0.816 | 0.843 | 0.032 | 0.061 |
| K | 3.99 | 0.417 | 0.394 | −0.059 | 0.116 |
| O | 6.72 | 0.531 | 0.543 | 0.023 | 0.425 |
| BM4208 | 20.00 | 0.900 | 0.906 | 0.007 | 0.616 |
| BM6506 | 10.57 | 0.712 | 0.806 | 0.116 | 0 |
| D | 12.75 | 0.745 | 0.818 | 0.089 | 0 |
| OarFCB | 13.76 | 0.846 | 0.806 | −0.051 | 0.004 |
| P | 9.96 | 0.697 | 0.816 | 0.146 | 0 |
| R | 3.00 | 0.442 | 0.430 | −0.027 | 0.505 |
| Mean | 10.69 | 0.674 | 0.711 | 0.052 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latch, E.K.; Gee, K.L.; Webb, S.L.; Honeycutt, R.L.; DeYoung, R.W.; Gonzales, R.A.; Demarais, S.; Toby, R. Genetic Consequences of Fence Confinement in a Population of White-Tailed Deer. Diversity 2021, 13, 126. https://doi.org/10.3390/d13030126
Latch EK, Gee KL, Webb SL, Honeycutt RL, DeYoung RW, Gonzales RA, Demarais S, Toby R. Genetic Consequences of Fence Confinement in a Population of White-Tailed Deer. Diversity. 2021; 13(3):126. https://doi.org/10.3390/d13030126
Chicago/Turabian StyleLatch, Emily K., Kenneth L. Gee, Stephen L. Webb, Rodney L. Honeycutt, Randy W. DeYoung, Robert A. Gonzales, Stephen Demarais, and Ryan Toby. 2021. "Genetic Consequences of Fence Confinement in a Population of White-Tailed Deer" Diversity 13, no. 3: 126. https://doi.org/10.3390/d13030126
APA StyleLatch, E. K., Gee, K. L., Webb, S. L., Honeycutt, R. L., DeYoung, R. W., Gonzales, R. A., Demarais, S., & Toby, R. (2021). Genetic Consequences of Fence Confinement in a Population of White-Tailed Deer. Diversity, 13(3), 126. https://doi.org/10.3390/d13030126