
Rice Plant–Soil Microbiome Interactions Driven by Root and Shoot Biomass

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Rice Genotype Selection and Experimental Design
2.2. Plant and Soil Sampling and DNA Extraction
2.3. Shotgun Metagenomic Library Construction and Illumina Sequencing
2.4. Sequence Processing
2.5. Community and Multivariate Statistical Analyses (PCoA and PLS)
3. Results
3.1. Selection of Recombinant Inbred Lines Segregating for Root and Shoot Biomass
3.2. Whole Microbial Community Structure and Impact of Shoot and Root Biomass Traits
3.3. Shoot and Root Biomass Driven Species Level Analysis
3.4. Rhizosphere Soil Microbial Community Function Analysis
3.5. Metagenomic Gene Level Analysis With PLS
4. Discussion
4.1. Soil Microbial Populations Associated With Rice Shoot and Root Biomass
4.2. Microbial Community Functions Correlated to Biomass Traits
4.3. Gene Trends Related to Shoot and Root Biomass
4.4. Relationship of Developmental Stage to Microbial Community Structure and Functions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friesen, M.L.; Porter, S.S.; Stark, S.C.; Von Wettberg, E.J.; Sachs, J.L.; Martinez-Romero, E. Microbially Mediated Plant Functional Traits. Annu. Rev. Ecol. Evol. Syst. 2011, 42, 23–46. [Google Scholar] [CrossRef] [Green Version]
- Berg, G.; Smalla, K. Plant species and soil type cooperatively shape the structure and function of microbial commu-nities in the rhizosphere. FEMS Microbiol. Ecol. 2009, 68, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudoin, E.; Benizri, E.; Guckert, A. Impact of growth stage on the bacterial community structure along maize roots, as determined by metabolic and genetic fingerprinting. Appl. Soil Ecol. 2002, 19, 135–145. [Google Scholar] [CrossRef]
- Inceoğlu, O.; Salles, J.F.; van Overbeek, L.; van Elsas, J.D. Effects of plant genotype and growth stage on the betaproteo-bacterial communities associated with different potato cultivars in two fields. Appl. Environ. Microbiol. 2010, 76, 3675–3684. [Google Scholar] [CrossRef] [Green Version]
- Van Overbeek, L.; Van Elsas, J.D. Effects of plant genotype and growth stage on the structure of bacterial communi-ties associated with potato (Solanum tuberosum L.). FEMS Microbiol. Ecol. 2008, 64, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; Van Der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Genet. 2013, 11, 789–799. [Google Scholar] [CrossRef]
- Santos-Medellín, C.; Edwards, J.; Liechty, Z.; Nguyen, B.; Sundaresan, V. Drought Stress Results in a Compartment-Specific Restructuring of the Rice Root-Associated Microbiomes. mBio 2017, 8, e00764-17. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.A.; Santos-Medellín, C.M.; Liechty, Z.S.; Nguyen, B.; Lurie, E.; Eason, S.; Phillips, G.; Sundaresan, V. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018, 16, e2003862. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [Green Version]
- Okubo, T.; Tokida, T.; Ikeda, S.; Bao, Z.; Tago, K.; Hayatsu, M.; Nakamura, H.; Sakai, H.; Usui, Y.; Hayashi, K.; et al. Effects of Elevated Carbon Dioxide, Elevated Temperature, and Rice Growth Stage on the Community Structure of Rice Root–Associated Bacteria. Microbes Environ. 2014, 29, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knief, C.; Delmotte, N.; Chaffron, S.; Stark, M.; Innerebner, G.; Wassmann, R.; Von Mering, C.; A Vorholt, J. Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J. 2012, 6, 1378–1390. [Google Scholar] [CrossRef] [Green Version]
- Hussain, Q.; Pan, G.; Liu, Y.; Zhang, A.; Li, L.; Zhang, X.; Jin, Z. Microbial community dynamics and function associated with rhizosphere over periods of rice growth. Plant Soil Environ. 2012, 58, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Liu, Q.; Li, Z.; Cheng, W.; Sun, J.; Guo, Z.; Li, Y.; Zhou, J.; Meng, D.; Li, H.; et al. Environmental factors shaping the diversity of bacterial communities that promote rice production. BMC Microbiol. 2018, 18, 51. [Google Scholar] [CrossRef] [Green Version]
- Breidenbach, B.; Pump, J.; Dumont, M.G. Microbial Community Structure in the Rhizosphere of Rice Plants. Front. Microbiol. 2016, 6, 1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knief, C.; Delmotte, N.; Vorholt, J.A. Bacterial adaptation to life in association with plants A proteomic perspective from culture to in situ conditions. Proteomics 2011, 11, 3086–3105. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Wang, F.; Zhang, J.; Chen, Y.; Zhang, C.; Liu, G.; Zhang, H.; Ma, C.; Zhang, J. The Variation in the Rhizosphere Microbiome of Cotton with Soil Type, Genotype and Developmental Stage. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nat. Cell Biol. 2012, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Veach, A.M.; Morris, R.; Yip, D.Z. Rhizosphere microbiomes diverge among Populus trichocarpa plant–host gen-otypes and chemotypes, but it depends on soil origin. Microbiome 2019, 7, 76. [Google Scholar] [CrossRef]
- Breidenbach, B.; Econrad, R. Seasonal dynamics of bacterial and archaeal methanogenic communities in flooded rice fields and effect of drainage. Front. Microbiol. 2015, 5, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, S.; Mitter, B.; Oswald, A.; Schloter-Hai, B.; Schloter, M.; Declerck, S.; Sessitsch, A. Rhizosphere microbiomes of potato cultivated in the High Andes show stable and dynamic core microbiomes with different responses to plant development. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, I.F. Tansley Review No. 27 The control of carbon partitioning in plants. New Phytol. 1990, 116, 341–381. [Google Scholar] [CrossRef]
- Briones, A.M.; Okabe, S.; Umemiya, Y.; Ramsing, N.-B.; Reichardt, W.; Okuyama, H. Influence of Different Cultivars on Populations of Ammonia-Oxidizing Bacteria in the Root Environment of Rice. Appl. Environ. Microbiol. 2002, 68, 3067–3075. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [Green Version]
- Aira, M.; Gómez-Brandón, M.; Lazcano, C.; Bååth, E.; Domínguez, J. Plant genotype strongly modifies the structure and growth of maize rhizosphere microbial communities. Soil Biol. Biochem. 2010, 42, 2276–2281. [Google Scholar] [CrossRef]
- Bouffaud, M.; Kyselková, M.; Gouesnard, B.; Grundmann, G.; Muller, D.; Moënne-Loccoz, Y. Is diversification history of maize influencing selection of soil bacteria by roots? Mol. Ecol. 2011, 21, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.-X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X.; et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, M.G.A.; Klironomos, J.N.; Ursic, M.; Moutoglis, P.; Streitwolf-Engel, R.; Boller, T.; Wiemken, A.; Sanders, I.R. Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nat. Cell Biol. 1998, 396, 69–72. [Google Scholar] [CrossRef]
- Wardle, D.A.; Bardgett, R.D.; Klironomos, J.N.; Setälä, H.; Van Der Putten, W.H.; Wall, D.H. Ecological Linkages between Aboveground and Belowground Biota. Science 2004, 304, 1629–1633. [Google Scholar] [CrossRef] [PubMed]
- Leff, J.W.; Bardgett, R.D.; Wilkinson, A. Predicting the structure of soil communities from plant community tax-onomy, phylogeny, and traits. ISME J. 2018, 12, 1794–1805. [Google Scholar] [CrossRef] [Green Version]
- Paliy, O.; Shankar, V. Application of multivariate statistical techniques in microbial ecology. Mol. Ecol. 2016, 25, 1032–1057. [Google Scholar] [CrossRef] [Green Version]
- Ullah, A.; Akbar, A.; Luo, Q. Microbiome Diversity in Cotton Rhizosphere under Normal and Drought Condi-tions. Microb. Ecol. 2019, 77, 429–439. [Google Scholar] [CrossRef]
- Ma, J.; Tang, J.Y.; Wang, S. Illumina sequencing of bacterial 16S rDNA and 16S rRNA reveals seasonal and spe-cies–specific variation in bacterial communities in four moss species. Appl. Microbiol. Biotechnol. 2017, 101, 6739–6753. [Google Scholar] [CrossRef]
- Dang, P.; Gao, Y.; Liu, J.; Yu, S.; Zhao, Z. Effects of thinning intensity on understory vegetation and soil microbial communities of a mature Chinese pine plantation in the Loess Plateau. Sci. Total. Environ. 2018, 630, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Counce, P.A.; Keisling, T.C.; Mitchell, A.J. A Uniform, Objective, and Adaptive System for Expressing Rice Development. Crop Sci. 2000, 40, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Kepler, R.M.; Schmidt, D.J.E.; Yarwood, S.A.; Cavigelli, M.A.; Reddy, K.N.; Duke, S.O.; Bradley, C.A.; Williams, M.M.; Buyer, J.S.; Maul, J.E. Soil Microbial Communities in Diverse Agroecosystems Exposed to the Herbicide Glyphosate. Appl. Environ. Microbiol. 2020, 86, e01744-19. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, A.J.; Blanchet, F.G.; Kindt, R. Vegan: Community Ecology Package Version 2.5-7 2020. Available online: http://CRAN.R-project.org/package=vegan (accessed on 1 February 2021).
- Lee, L.C.; Liong, C.-Y.; Jemain, A.A. Partial least squares-discriminant analysis (PLS-DA) for classification of high-dimensional (HD) data: A review of contemporary practice strategies and knowledge gaps. Analysis 2018, 143, 3526–3539. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS–regression: A basic tool of chemometrics. Chemom Intell Lab Syst 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Wold, S.; Geladi, P.; Esbensen, K.; Öhman, J. Multi-way principal components-and PLS-analysis. J. Chemom. 1987, 1, 41–56. [Google Scholar] [CrossRef]
- Lingens, F.; Blecher, R.; Blecher, H.; Blobel, F.; Eberspächer, J.; Fröhner, C.; Gorisch, H.; Layh, G. Phenylobacterium immobile gen. nov., sp. nov., a Gram-Negative Bacterium That Degrades the Herbicide Chloridazon. Int. J. Syst. Bacteriol. 1985, 35, 26–39. [Google Scholar] [CrossRef]
- Abraham, W.-R.; Macedo, A.J.; Lunsdorf, H. Phylogeny by a polyphasic approach of the order Caulobacterales, proposal of Caulobacter mirabilis sp. nov., Phenylobacterium haematophilum sp. nov. and Phenylobacterium conjunc-tum sp. nov., and emendation of the genus Phenylobacterium. Int. J. Syst. Evol. Microbiol. 2008, 58, 1939–1949. [Google Scholar] [CrossRef]
- Ding, L.-J.; Cui, H.-L.; Nie, S.-A.; Long, X.-E.; Duan, G.-L.; Zhu, Y.-G. Microbiomes inhabiting rice roots and rhizosphere. FEMS Microbiol. Ecol. 2019, 95, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ishii, S.; Ikeda, S.; Minamisawa, K.; Senoo, K. Nitrogen Cycling in Rice Paddy Environments: Past Achievements and Future Challenges. Microbes Environ. 2011, 26, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Hirano, K.; Sugiyama, T.; Kosugi, A.; Nioh, I.; Asai, T.; Nakai, H. Relationship between number of nitrogen–fixing rihzobacteria and growth pattern of rice varieties in the nature farming. Breed Res. 2001, 3, 3–12. [Google Scholar] [CrossRef]
- Sanford, R.A.; Cole, J.R.; Tiedje, J.M.; Tiedje, J.M. Characterization and description of Anaeromyxobacter dehalogenans gen. nov., sp. nov., an aryl–halorespiring facultative anaerobic myxobacterium. Appl. Environ. Microbiol. 2002, 68, 893–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Kim, S.Y.; Kim, P.J.; Madsen, E.L.; Jeon, C.O. Methane emission and dynamics of methanotrophic and methanogenic communities in a flooded rice field ecosystem. FEMS Microbiol. Ecol. 2014, 88, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Neue, H.-U. Methane Emission from Rice Fields. Bioscience 1993, 43, 466–474. [Google Scholar] [CrossRef]
- Sauder, L.A.; Albertsen, M.; Engel, K.; Schwarz, J.; Nielsen, P.H.; Wagner, M.; Neufeld, J.D. Cultivation and characterization of Candidatus Nitrosocosmicus exaquare, an ammonia-oxidizing archaeon from a municipal wastewater treatment system. ISME J. 2017, 11, 1142–1157. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.-Y.; Kim, J.-G.; Sinninghe Damsté, J.S. A hydrophobic ammonia–oxidizing archaeon of the Nitrosocosmicus clade isolated from coal tar–contaminated sediment. Environ. Microbiol. Rep. 2016, 8, 983–992. [Google Scholar] [CrossRef]
- Kirk, G.J.D.; Kronzucker, H.J. The Potential for Nitrification and Nitrate Uptake in the Rhizosphere of Wetland Plants: A Modelling Study. Ann. Bot. 2005, 96, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Kirk, G. Plant-mediated processess to acquire nutrients: Nitrogen uptake by rice plants. Plant Soil 2001, 232, 129–134. [Google Scholar] [CrossRef]
- Park, B.B.; Yanai, R.D.; Vadeboncoeur, M.A.; Hamburg, S.P. Estimating Root Biomass in Rocky Soils using Pits, Cores, and Allometric Equations. Soil Sci. Soc. Am. J. 2007, 71, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Pandey, G.; Jain, R.K. Bacterial Chemotaxis toward Environmental Pollutants: Role in Bioremediation. Appl. Environ. Microbiol. 2002, 68, 5789–5795. [Google Scholar] [CrossRef] [Green Version]
- Philippot, L. Denitrifying genes in bacterial and Archaeal genomes. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 2002, 1577, 355–376. [Google Scholar] [CrossRef]
- Kuzyakov, Y.; Domanski, G. Carbon input by plants into the soil. Review. J. Plant Nutr. Soil Sci. 2000, 163, 421–431. [Google Scholar] [CrossRef]
- Mougel, C.; Offre, P.; Ranjard, L. Dynamic of the genetic structure of bacterial and fungal communities at differ-ent developmental stages of Medicago truncatula Gaertn. cv. Jemalong line J5. New Phytol. 2006, 170, 165–175. [Google Scholar] [CrossRef]
- Mayer, E.; Dörr de Quadros, P.; Fulthorpe, R. Plantibacter flavus, Curtobacterium herbarum, Paenibacillus tai-chungensis, and Rhizobium selenitireducens Endophytes Provide Host–Specific Growth Promotion of Arabidopsis tha-liana, Basil, Lettuce, and Bok Choy Plants. Appl. Environ. Microbiol. 2019, 85, e00383-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
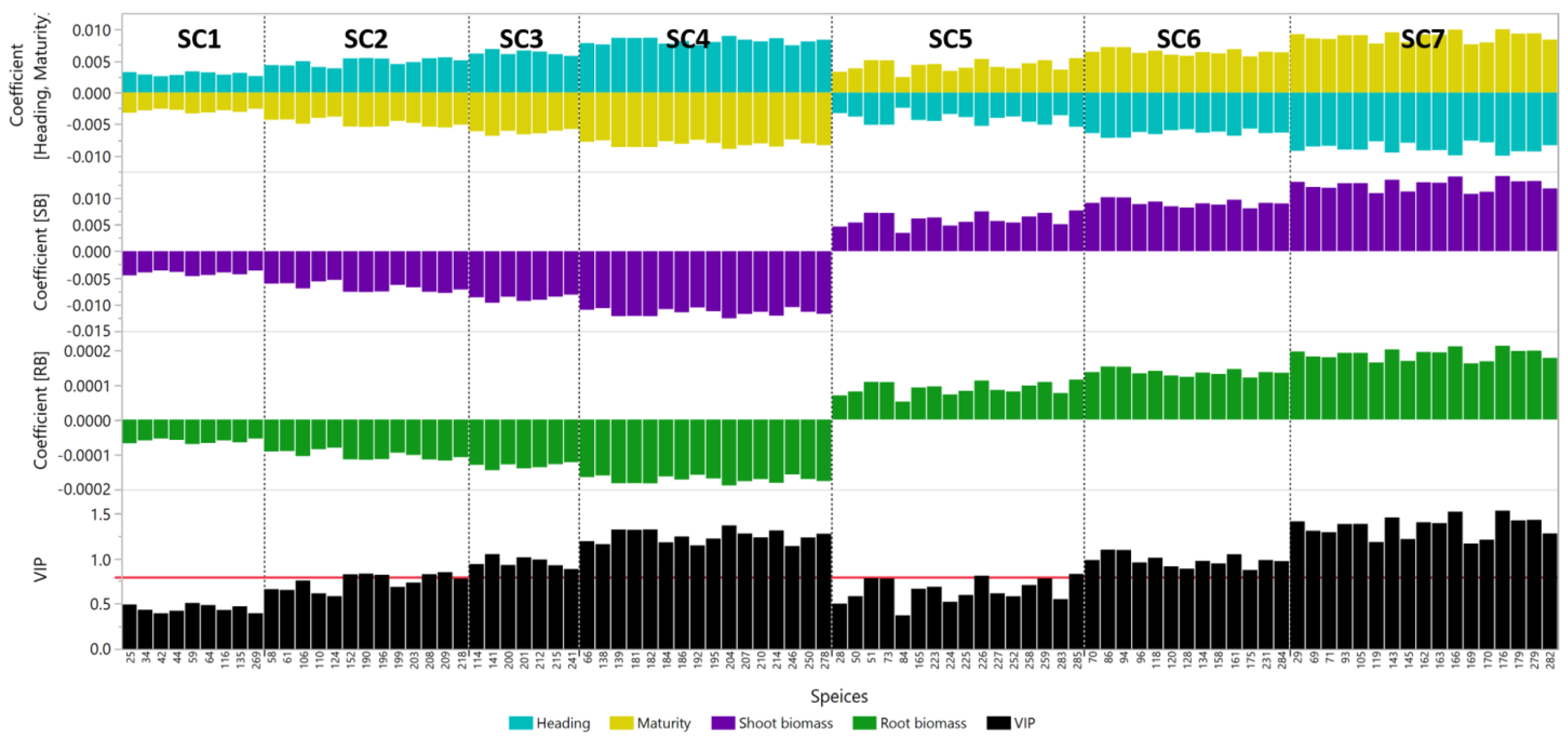
| Species Cluster | Species Name | ID | SB Coefficient | RB Coefficient | VIP |
|---|---|---|---|---|---|
| SC1 | Acidobacteria bacterium 13_2_20CM_2_66_4 | 25 | −4.5 × 10−3 | −6.8 × 10−5 | 0.491 |
| Acidobacteria bacterium RIFCSPLOWO2_12_FULL_66_10 | 34 | −4.0 × 10−3 | −6.0 × 10−5 | 0.433 | |
| Niastella koreensis | 42 | −3.6 × 10−3 | −5.4 × 10−5 | 0.393 | |
| Bacteriodetes bacterium 13_1_20CM_4_60_6 | 44 | −3.9 × 10−3 | −5.8 × 10−5 | 0.422 | |
| Gemmatimonadetes bacterium | 59 | −4.7 × 10−3 | −7.0 × 10−5 | 0.508 | |
| Gemmatimonadetes bacterium SCN 70−22 | 64 | −4.5 × 10−3 | −6.7 × 10−5 | 0.484 | |
| Cyanobacteria bacterium 13_1_20CM_4_61_6 | 269 | −3.6 × 10−3 | −5.5 × 10−5 | 0.395 | |
| Betaproteobacteria bacterium RIFCSPLOWO2_12_FULL_62_13 | 116 | −4.0 × 10−3 | −6.0 × 10−5 | 0.431 | |
| Deltaproteobacteria bacterium RIFCSPLOWO2_12_FULL_60_19 | 135 | −4.4 × 10−3 | −6.5 × 10−5 | 0.471 | |
| SC2 | Rhizobacter sp. Root404 | 110 | −5.7 × 10−3 | −8.5 × 10−5 | 0.614 |
| Hyalangium minutum | 124 | −5.4 × 10−3 | −8.1 × 10−5 | 0.583 | |
| Singulisphaera sp. GP187 | 152 | −7.6 × 10−3 | −1.1 × 10−4 | 0.824 | |
| Gemmatirosa kalamazoonesis | 58 | −6.1 × 10−3 | −9.1 × 10−5 | 0.660 | |
| Gemmatimonadetes bacterium 21-71-4 | 61 | −6.0 × 10−3 | −9.0 × 10−5 | 0.652 | |
| Alphaproteobacteria bacterium 64-11 | 106 | −7.0 × 10−3 | −1.0 × 10−4 | 0.756 | |
| Dactylosporangium aurantiacum | 190 | −7.7 × 10−3 | −1.2 × 10−4 | 0.832 | |
| Gaiella sp. SCGC AG-212-M14 | 196 | −7.6 × 10−3 | −1.1 × 10−4 | 0.819 | |
| Solirubrobacter soli | 199 | −6.3 × 10−3 | −9.5 × 10−5 | 0.686 | |
| Solirubrobacterales bacterium URHD0059 | 203 | −6.8 × 10−3 | −1.0 × 10−4 | 0.733 | |
| Actinobacteria bacterium 13_1_20CM_4_68_12 | 208 | −7.6 × 10−3 | −1.1 × 10−4 | 0.826 | |
| Actinobacteria bacterium 13_1_20CM_4_69_9 | 209 | −7.8 × 10−3 | −1.2 × 10−4 | 0.848 | |
| Fimbriimonas ginsengisoli | 218 | −7.2 × 10−3 | −1.1 × 10−4 | 0.778 | |
| SC3 | Betaproteobacteria bacterium GR16-43 | 114 | −8.7 × 10−3 | −1.3 × 10−4 | 0.938 |
| Rudaea cellulosilytica | 141 | −9.7 × 10−3 | −1.5 × 10−4 | 1.048 | |
| Solirubrobacter sp. URHD0082 | 200 | −8.6 × 10−3 | −1.3 × 10−4 | 0.927 | |
| Solirubrobacterales bacterium 67−14 | 201 | −9.3 × 10−3 | −1.4 × 10−4 | 1.012 | |
| Actinobacteria bacterium 13_2_20CM_68_14 | 212 | −9.1 × 10−3 | −1.4 × 10−4 | 0.988 | |
| Actinobacteria bacterium RBG_16_68_12 | 215 | −8.5 × 10−3 | −1.3 × 10−4 | 0.924 | |
| Kouleothrix aurantiaca | 241 | −8.2 × 10−3 | −1.2 × 10−4 | 0.883 | |
| SC4 | Phycicoccus cremeus | 184 | −1.1 × 10−2 | −1.6 × 10−4 | 1.178 |
| Phenylobacterium sp. RIFCSPHIGHO2_01_FULL_69_31 | 66 | −1.1 × 10−2 | −1.6 × 10−4 | 1.192 | |
| Gammaproteobacteria bacterium RIFCSPHIGHO2_12_FULL_63_22 | 138 | −1.1 × 10−2 | −1.6 × 10−4 | 1.159 | |
| Dokdonella immobilis | 139 | −1.2 × 10−2 | −1.8 × 10−4 | 1.319 | |
| Jatrophihabitans endophyticus | 181 | −1.2 × 10−2 | −1.8 × 10−4 | 1.316 | |
| Kineosporia sp. A_224 | 182 | −1.2 × 10−2 | −1.8 × 10−4 | 1.321 | |
| Actinoplanes awajinensis | 186 | −1.1 × 10−2 | −1.7 × 10−4 | 1.242 | |
| Nocardioides halotolerans | 192 | −1.1 × 10−2 | −1.6 × 10−4 | 1.144 | |
| Actinobacteria bacterium IMCC26256 | 195 | −1.1 × 10−2 | −1.7 × 10−4 | 1.220 | |
| Thermoleophilum album | 204 | −1.3 × 10−2 | −1.9 × 10−4 | 1.364 | |
| Actinobacteria bacterium 13_1_20CM_3_71_11 | 207 | −1.2 × 10−2 | −1.8 × 10−4 | 1.275 | |
| Actinobacteria bacterium 13_2_20CM_2_66_6 | 210 | −1.1 × 10−2 | −1.7 × 10−4 | 1.233 | |
| Actinobacteria bacterium RBG_16_67_10 | 214 | −1.2 × 10−2 | −1.8 × 10−4 | 1.309 | |
| Chloroflexi bacterium 13_1_40CM_4_68_4 | 246 | −1.1 × 10−2 | −1.6 × 10−4 | 1.137 | |
| Chloroflexi bacterium GWC2_73_18 | 250 | −1.1 × 10−2 | −1.7 × 10−4 | 1.231 | |
| bacterium JGI 053 | 278 | −1.2 × 10−2 | −1.8 × 10−4 | 1.272 | |
| SC5 | Bacteroidetes bacterium RBG_13_42_15 | 50 | 5.4 × 10−3 | 8.1 × 10−5 | 0.583 |
| Bacteroidetes bacterium RBG_13_43_22 | 51 | 7.2 × 10−3 | 1.1 × 10−4 | 0.781 | |
| Rhodopseudomonas palustris | 73 | 7.2 × 10−3 | 1.1 × 10−4 | 0.778 | |
| Pseudorhodoplanes sinuspersici | 84 | 3.4 × 10−3 | 5.1 × 10−5 | 0.372 | |
| Planctomycetes bacterium RBG_16_55_9 | 165 | 6.1 × 10−3 | 9.2 × 10−5 | 0.665 | |
| Anaerolinea thermophila | 223 | 6.3 × 10−3 | 9.5 × 10−5 | 0.687 | |
| Bellilinea caldifistulae | 224 | 4.8 × 10−3 | 7.2 × 10−5 | 0.521 | |
| Leptolinea tardivitalis | 225 | 5.5 × 10−3 | 8.2 × 10−5 | 0.596 | |
| Levilinea saccharolytica | 226 | 7.5 × 10−3 | 1.1 × 10−4 | 0.809 | |
| Longilinea arvoryzae | 227 | 5.7 × 10−3 | 8.5 × 10−5 | 0.614 | |
| Chloroflexi bacterium HGW-Chloroflexi-10 | 252 | 5.4 × 10−3 | 8.1 × 10−5 | 0.583 | |
| Chloroflexi bacterium RBG_16_54_18 | 258 | 6.5 × 10−3 | 9.8 × 10−5 | 0.706 | |
| Chloroflexi bacterium RBG_16_57_11 | 259 | 7.2 × 10−3 | 1.1 × 10−4 | 0.779 | |
| Candidatus Nitrososphaera evergladensis | 283 | 5.1 × 10−3 | 7.6 × 10−5 | 0.550 | |
| Oxytricha trifallax | 285 | 7.7 × 10−3 | 1.1 × 10−4 | 0.828 | |
| Acidobacteria bacterium RBG_13_68_16 | 28 | 4.6 × 10−3 | 6.9 × 10−5 | 0.500 | |
| SC6 | Pseudolabrys sp. Root1462 | 86 | 1.0 × 10−2 | 1.5 × 10−4 | 1.097 |
| Bradyrhizobium erythrophlei | 70 | 9.1 × 10−3 | 1.4 × 10−4 | 0.983 | |
| Rhodospirillales bacterium 69-11 | 94 | 1.0 × 10−2 | 1.5 × 10−4 | 1.094 | |
| Rhodospirillales bacterium URHD0088 | 96 | 8.8 × 10−3 | 1.3 × 10−4 | 0.956 | |
| Anaeromyxobacter dehalogenans | 118 | 9.3 × 10−3 | 1.4 × 10−4 | 1.008 | |
| Anaeromyxobacter sp. RBG_16_69_14 | 120 | 8.4 × 10−3 | 1.3 × 10−4 | 0.913 | |
| Labilithrix luteola | 128 | 8.2 × 10−3 | 1.2 × 10−4 | 0.886 | |
| Myxococcales bacterium 68-20 | 134 | 9.0 × 10−3 | 1.3 × 10−4 | 0.973 | |
| Planctomycetes bacterium GWF2_41_51 | 158 | 8.7 × 10−3 | 1.3 × 10−4 | 0.945 | |
| Planctomycetes bacterium RBG_13_50_24 | 161 | 9.7 × 10−3 | 1.4 × 10−4 | 1.047 | |
| Anaerolineae bacterium CG2_30_64_16 | 231 | 9.1 × 10−3 | 1.4 × 10−4 | 0.983 | |
| Candidatus Nitrososphaera gargensis | 284 | 9.0 × 10−3 | 1.3 × 10−4 | 0.970 | |
| Verrucomicrobia bacterium 13_2_20CM_54_12 | 175 | 8.1 × 10−3 | 1.2 × 10−4 | 0.872 | |
| SC7 | Alphaproteobacteria bacterium 13_2_20CM_2_64_7 | 105 | 1.3 × 10−2 | 1.9 × 10−4 | 1.382 |
| Phycisphaerae bacterium SG8_4 | 143 | 1.3 × 10−2 | 2.0 × 10−4 | 1.452 | |
| Verrucomicrobia bacterium 13_1_20CM_4_54_11 | 170 | 1.1 × 10−2 | 1.7 × 10−4 | 1.207 | |
| Verrucomicrobia bacterium 13_2_20CM_55_10 | 176 | 1.4 × 10−2 | 2.1 × 10−4 | 1.528 | |
| Methanocella arvoryzae | 279 | 1.3 × 10−2 | 2.0 × 10−4 | 1.427 | |
| Candidatus Nitrosocosmicus oleophilus | 282 | 1.2 × 10−2 | 1.8 × 10−4 | 1.276 | |
| Acidobacteria bacterium RBG_16_70_10 | 29 | 1.3 × 10−2 | 1.9 × 10−4 | 1.409 | |
| Bradyrhizobium elkanii | 69 | 1.2 × 10−2 | 1.8 × 10−4 | 1.306 | |
| Bradyrhizobium japonicum | 71 | 1.2 × 10−2 | 1.8 × 10−4 | 1.290 | |
| Rhodospirillales bacterium 20-64-7 | 93 | 1.3 × 10−2 | 1.9 × 10−4 | 1.381 | |
| Anaeromyxobacter sp. Fw109-5 | 119 | 1.1 × 10−2 | 1.6 × 10−4 | 1.182 | |
| Phycisphaerae bacterium SM23_33 | 145 | 1.1 × 10−2 | 1.7 × 10−4 | 1.214 | |
| Planctomycetes bacterium RBG_13_60_9 | 162 | 1.3 × 10−2 | 1.9 × 10−4 | 1.400 | |
| Planctomycetes bacterium RBG_13_62_9 | 163 | 1.3 × 10−2 | 1.9 × 10−4 | 1.391 | |
| Planctomycetes bacterium RBG_16_64_12 | 166 | 1.4 × 10−2 | 2.1 × 10−4 | 1.518 | |
| Verrucomicrobia bacterium 13_1_20CM_3_54_17 | 169 | 1.1 × 10−2 | 1.6 × 10−4 | 1.164 | |
| Pedosphaera parvula | 179 | 1.3 × 10−2 | 2.0 × 10−4 | 1.421 |
| Gene Cluster | Gene | Gene ID | SB Coefficient | RB Coefficient | VIP |
|---|---|---|---|---|---|
| GC1 | 2–methylcitrate dehydratase FeS dependent (EC 4.2.1.79) | 23 | −0.008 | 0.018 | 0.98 |
| Arginine ABC transporter, permease protein ArtM | 36 | −0.010 | 0.027 | 1.46 | |
| Ornithine aminotransferase (EC 2.6.1.13) | 63 | −0.004 | 0.021 | 1.31 | |
| 3,5–diaminohexanoate dehydrogenase (EC 1.4.1.11) | 401 | −0.006 | 0.022 | 1.25 | |
| L–threonine transporter, anaerobically inducible | 555 | −0.011 | 0.017 | 1.38 | |
| 2–ketogluconate kinase (EC 2.7.1.13) | 626 | −0.012 | 0.029 | 1.55 | |
| Maltose operon transcriptional repressor MalR, LacI family | 1439 | −0.007 | 0.024 | 1.34 | |
| Putative regulator of the mannose operon, ManO | 1509 | −0.010 | 0.023 | 1.23 | |
| poly(beta–D–mannuronate) lyase (EC 4.2.2.3) | 1899 | −0.007 | 0.023 | 1.19 | |
| Substrate–specific component YkoE of thiamin–regulated ECF transporter for HydroxyMethylPyrimidine | 2442 | −0.008 | 0.014 | 0.91 | |
| DNA polymerase–like protein MT3142 | 2904 | −0.010 | 0.016 | 1.12 | |
| Polyketide beta–ketoacyl synthase WhiE–KS paralog | 3131 | −0.008 | 0.016 | 0.91 | |
| Phytoene desaturase, neurosporene or lycopene producing (EC 1.3.–.–) | 3246 | −0.011 | 0.019 | 1.20 | |
| Fatty acyl–coenzyme A elongase | 3263 | −0.009 | 0.027 | 1.42 | |
| Acyl carrier protein (ACP1) | 3331 | −0.011 | 0.029 | 1.51 | |
| FIG027190: Putative transmembrane protein | 3338 | −0.007 | 0.020 | 1.06 | |
| Triacylglycerol lipase precursor (EC 3.1.1.3) | 3372 | −0.009 | 0.012 | 0.97 | |
| UPF0225 protein YchJ | 3385 | −0.008 | 0.021 | 1.08 | |
| Haemin uptake system permease protein | 3422 | −0.011 | 0.024 | 1.40 | |
| Probable Lysine n(6)–hydroxylase associated with siderophore S biosynthesis (EC 1.14.13.59) | 3502 | −0.006 | 0.020 | 1.10 | |
| Dipeptide transport system permease protein DppC (TC 3.A.1.5.2) | 3605 | −0.004 | 0.019 | 1.27 | |
| Transcriptional regulator of fimbriae expression FimZ (LuxR/UhpA family) | 3791 | −0.012 | 0.020 | 1.41 | |
| Phenylacetaldehyde dehydrogenase (EC 1.2.1.39) | 3929 | −0.007 | 0.019 | 0.99 | |
| Vanillate O–demethylase oxygenase subunit (EC 1.14.13.82) | 4059 | −0.005 | 0.018 | 0.99 | |
| Protein gp47, recombination–related [Bacteriophage A118] | 4705 | −0.006 | 0.020 | 1.10 | |
| Uncharacterized transporter, similarity to citrate transporter | 4969 | −0.008 | 0.015 | 0.88 | |
| SSU ribosomal protein S10p (S20e), chloroplast | 5339 | −0.006 | 0.020 | 1.10 | |
| SSU ribosomal protein S13p (S18e), mitochondrial | 5382 | −0.009 | 0.029 | 1.53 | |
| Putative succinate dehydrogenase cytochrome b subunit | 6059 | −0.007 | 0.014 | 0.83 | |
| Sigma factor RpoE negative regulatory protein RseB precursor | 6373 | −0.006 | 0.025 | 1.46 | |
| tRNA methylase YGL050w homolog Wyeosine biosynthesis | 6399 | −0.006 | 0.020 | 1.10 | |
| Diaminobutyrate–pyruvate aminotransferase (EC 2.6.1.46) | 6476 | −0.008 | 0.025 | 1.33 | |
| Glutaredoxin 1 | 6486 | −0.010 | 0.022 | 1.26 | |
| RsbS, negative regulator of sigma–B | 6603 | −0.005 | 0.028 | 1.72 | |
| GC2 | (GlcNAc)2 ABC transporter, permease component 2 | 621 | −0.015 | 0.035 | 1.94 |
| Alpha–N–acetylglucosaminidase (EC 3.2.1.50) | 811 | −0.012 | 0.039 | 2.06 | |
| Cyanate ABC transporter, ATP–binding protein | 4406 | −0.012 | 0.041 | 2.23 | |
| Phage capsid and scaffold | 4639 | −0.012 | 0.038 | 1.99 | |
| Autoinducer 2 (AI–2) ABC transport system, membrane channel protein LsrC | 5486 | −0.018 | 0.043 | 2.38 | |
| High–affinity choline uptake protein BetT | 6461 | −0.013 | 0.032 | 1.71 | |
| GC8 | Arginine pathway regulatory protein ArgR, repressor of arg regulon | 44 | 0.003 | −0.014 | 0.99 |
| Acetyl–CoA acetyltransferase (EC 2.3.1.9) | 406 | 0.004 | −0.013 | 1.00 | |
| Methionyl–tRNA formyltransferase (EC 2.1.2.9) | 2587 | 0.006 | −0.016 | 1.19 | |
| O–succinylbenzoate synthase (EC 4.2.1.113) | 2616 | 0.003 | −0.014 | 1.05 | |
| Phosphoribosylaminoimidazole carboxylase catalytic subunit (EC 4.1.1.21) | 4527 | 0.004 | −0.014 | 1.02 | |
| Pyridine nucleotide–disulphide oxidoreductase associated with reductive pyrimidine catabolism | 4616 | 0.004 | −0.018 | 1.19 | |
| Glutamyl–tRNA(Gln) amidotransferase subunit B (EC 6.3.5.7) | 4993 | 0.004 | −0.014 | 1.03 | |
| LSU ribosomal protein L5p (L11e) | 5240 | 0.005 | −0.011 | 1.04 | |
| UbiD family decarboxylase, MJ1133 type | 412 | 0.011 | −0.005 | 1.62 | |
| Lacto–N–biose phosphorylase (EC 2.4.1.211) | 1381 | 0.011 | −0.006 | 1.72 | |
| Putative DNA–binding protein in cluster with Type I restriction–modification system | 3047 | 0.013 | −0.011 | 1.68 | |
| Dipicolinate synthase subunit B | 3076 | 0.010 | −0.011 | 1.22 | |
| Spore germination protein GerKB | 3109 | 0.008 | −0.012 | 0.97 | |
| Stage IV sporulation protein A | 3177 | 0.013 | −0.005 | 1.96 | |
| LSU ribosomal protein L18e | 5267 | 0.009 | −0.012 | 1.07 | |
| LSU ribosomal protein L23Ae (L23p) | 5271 | 0.011 | −0.009 | 1.50 | |
| LSU ribosomal protein L30e | 5276 | 0.012 | −0.011 | 1.49 | |
| SSU ribosomal protein S27e | 5362 | 0.007 | −0.015 | 0.83 | |
| DNA–directed RNA polymerase II second largest subunit (EC 2.7.7.6) | 6261 | 0.007 | −0.015 | 0.84 | |
| GC10 | Meso–diaminopimelate D–dehydrogenase (EC 1.4.1.16) | 374 | 0.015 | −0.023 | 1.76 |
| S–adenosylmethionine decarboxylase proenzyme (EC 4.1.1.50), prokaryotic class 1A | 502 | 0.009 | −0.017 | 1.10 | |
| Predicted cellobiose ABC transport system, ATP–binding protein 1 | 764 | 0.010 | −0.032 | 1.74 | |
| Multiple sugar ABC transporter, substrate–binding protein | 1123 | 0.012 | −0.024 | 1.55 | |
| Predicted regulator of fructose utilization, DeoR family | 1138 | 0.011 | −0.022 | 1.27 | |
| Predicted L–rhamnose permease RhaY | 1341 | 0.010 | −0.022 | 1.23 | |
| Formylmethanofuran dehydrogenase (tungsten) operon gene G | 1526 | 0.010 | −0.020 | 1.17 | |
| Potassium uptake protein, integral membrane component, KtrB | 4938 | 0.012 | −0.015 | 1.40 | |
| Similar to ribosomal large subunit pseudouridine synthase D, CAC1266–type | 5215 | 0.009 | −0.026 | 1.35 | |
| SSU ribosomal protein S4p (S9e), mitochondrial | 5397 | 0.009 | −0.021 | 1.16 | |
| Signal peptidase, type IV – prepilin/preflagellin | 5413 | 0.012 | −0.017 | 1.35 | |
| Coenzyme F420H2 dehydrogenase (methanophenazine) subunit FpoM | 5845 | 0.013 | −0.022 | 1.48 | |
| Sulfhydrogenase II subunit g | 5959 | 0.014 | −0.027 | 1.64 | |
| Conjugative transfer protein TrbG | 6889 | 0.014 | −0.029 | 1.77 | |
| GC11 | IcmB (DotO) protein | 3646 | 0.017 | −0.038 | 2.12 |
| Possible alpha/beta hydrolase superfamily, slr1917 homolog | 4225 | 0.013 | −0.034 | 1.81 | |
| photosystem I subunit XI (PsaL) | 4833 | 0.015 | −0.037 | 2.01 | |
| Phycobilisome rod–core linker polypeptide, phycocyanin–associated | 4871 | 0.012 | −0.038 | 2.03 | |
| Conjugative signal peptidase TrhF | 6881 | 0.012 | −0.040 | 2.13 | |
| Inclusion membrane protein–52 | 6958 | 0.015 | −0.034 | 1.92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Baca, C.P.; Rivers, A.R.; Maul, J.E.; Kim, W.; Poudel, R.; McClung, A.M.; Roberts, D.P.; Reddy, V.R.; Barnaby, J.Y. Rice Plant–Soil Microbiome Interactions Driven by Root and Shoot Biomass. Diversity 2021, 13, 125. https://doi.org/10.3390/d13030125
Fernández-Baca CP, Rivers AR, Maul JE, Kim W, Poudel R, McClung AM, Roberts DP, Reddy VR, Barnaby JY. Rice Plant–Soil Microbiome Interactions Driven by Root and Shoot Biomass. Diversity. 2021; 13(3):125. https://doi.org/10.3390/d13030125
Chicago/Turabian StyleFernández-Baca, Cristina P., Adam R. Rivers, Jude E. Maul, Woojae Kim, Ravin Poudel, Anna M. McClung, Daniel P. Roberts, Vangimalla R. Reddy, and Jinyoung Y. Barnaby. 2021. "Rice Plant–Soil Microbiome Interactions Driven by Root and Shoot Biomass" Diversity 13, no. 3: 125. https://doi.org/10.3390/d13030125
APA StyleFernández-Baca, C. P., Rivers, A. R., Maul, J. E., Kim, W., Poudel, R., McClung, A. M., Roberts, D. P., Reddy, V. R., & Barnaby, J. Y. (2021). Rice Plant–Soil Microbiome Interactions Driven by Root and Shoot Biomass. Diversity, 13(3), 125. https://doi.org/10.3390/d13030125