


A High-Quality Genome Assembly of the Mitochondrial Genome of the Oil-Tea Tree Camellia gigantocarpa (Theaceae)



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Genome Sequencing
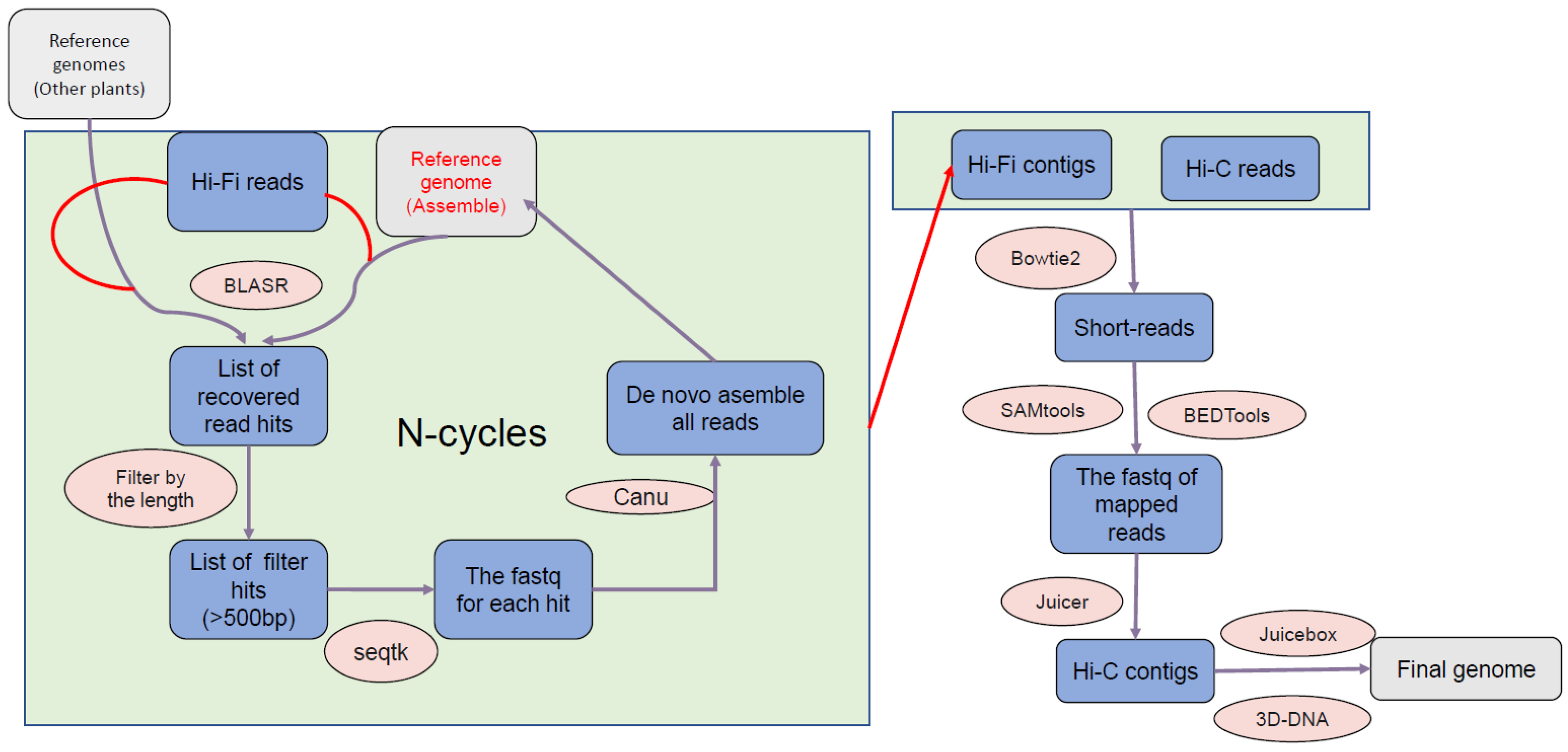
2.2. Genome Assembly
2.3. Genome Annotation and Visualization
2.4. Prediction of RNA-Editing Sites
2.5. Synteny Analysis
2.6. Phylogenetic Analysis
3. Results
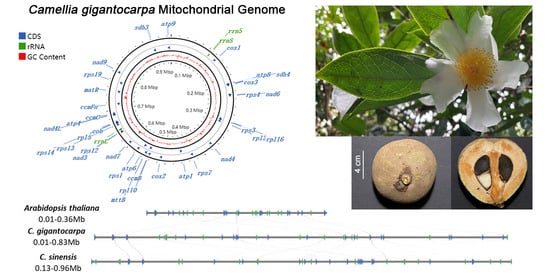
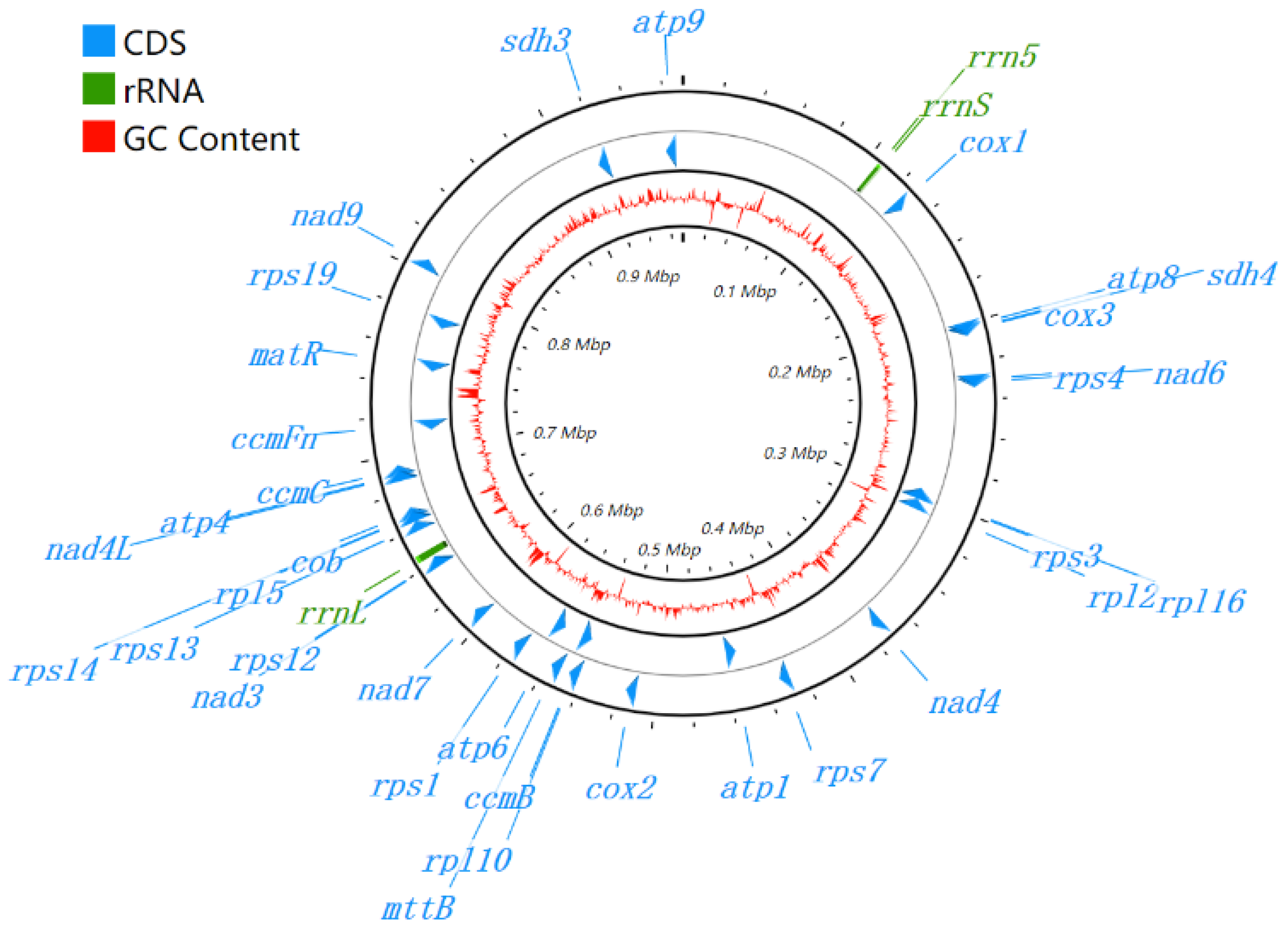
3.1. Genome Assembly and Genome Annotation
3.2. Identified Repetitive Sequences
3.3. The Prediction of RNA Editing
3.4. Comparison of the Genome Structure
3.5. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ye, Z.; Wu, Y.; Ul, H.M.Z.; Yan, W.; Yu, J.; Zhang, J.; Yao, G.; Hu, X. Complementary transcriptome and proteome profiling in the mature seeds of Camellia oleifera from Hainan island. PLoS ONE 2020, 15, e226888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieber, J.; Lindenmeyer, M.T.; Kampe, K.; Campbell, K.N.; Cohen, C.D.; Hopfer, H.; Mundel, P.; Jehle, A.W. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am. J. Physiol. Ren. Physiol. 2010, 299, F821–F829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.; Wang, K.; Zhou, C.; Xie, Y.; Yao, X.; Yin, H. Seed transcriptomics analysis in Camellia oleifera uncovers genes associated with oil content and fatty acid composition. Int. J. Mol. Sci. 2018, 19, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.F.; Huang, B.; Xu, M.; Zhou, P.Y.; Ni, Z.X.; Gong, C.; Wen, Q.; Cao, F.L.; Xu, L.A. The reference genome of Camellia chekiangoleosa provides insights into Camellia evolution and tea oil biosynthesis. Hortic. Res. 2022, 9, uhab083. [Google Scholar] [CrossRef]
- Li, J.; Luo, Z.; Zhang, C.; Qu, X.; Chen, M.; Song, T.; Yuan, J. Seasonal variation in the rhizosphere and non-rhizosphere microbial community structures and functions of Camellia yuhsienensis hu. Microorganisms 2020, 8, 1385. [Google Scholar] [CrossRef]
- Xie, Y. Fruit economic characters and seed oil components of seven kinds of oil-used Camellia. Chin. J. Trop. Crops 2016, 2, 427–431. [Google Scholar]
- Epstein, C.B.; Waddle, J.A.; Hale, W.; Davé, V.; Thornton, J.; Macatee, T.L.; Garner, H.R.; Butow, R.A. Genome-wide responses to mitochondrial dysfunction. Mol. Biol. Cell 2001, 12, 297–308. [Google Scholar] [CrossRef]
- Carafoli, E. The fateful encounter of mitochondria with calcium: How did it happen? Biochim. Biophys. Acta 2010, 1797, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Yu, F.; Tian, L.; Huang, X.; Tan, H.; Xie, Z.; Hao, X.; Li, D.; Luan, S.; Chen, L. RPS9M, a mitochondrial ribosomal protein, is essential for central cell maturation and endosperm development in Arabidopsis. Front. Plant Sci. 2017, 8, 2171. [Google Scholar] [CrossRef] [Green Version]
- Portereiko, M.F.; Sandaklie-Nikolova, L.; Lloyd, A.; Dever, C.A.; Otsuga, D.; Drews, G.N. NUCLEAR FUSION DEFECTIVE1 encodes the Arabidopsis RPL21M protein and is required for karyogamy during female gametophyte development and fertilization. Plant Physiol. 2006, 141, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Knoop, V. The mitochondrial DNA of land plants: Peculiarities in phylogenetic perspective. Curr. Genet. 2004, 46, 123–139. [Google Scholar] [CrossRef]
- Marechal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef]
- Yang, J.B.; Yang, S.X.; Li, H.T.; Yang, J.; Li, D.Z. Comparative chloroplast genomes of Camellia species. PLoS ONE 2013, 8, e73053. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, C.; Guo, X.; Liu, Q.; Wang, K. Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis. PLoS ONE 2019, 14, e216645. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Li, W.; Gao, C.; Zhang, D.; Gao, L. Deciphering tea tree chloroplast and mitochondrial genomes of Camellia sinensis var. assamica. Sci. Data 2019, 6, 209. [Google Scholar] [CrossRef] [Green Version]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.; Vert, J.; Heard, E.; Dekker, J.; Barillot, E. Hic-pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef] [Green Version]
- Kovar, L.; Nageswara-Rao, M.; Ortega-Rodriguez, S.; Dugas, D.V.; Straub, S.; Cronn, R.; Strickler, S.R.; Hughes, C.E.; Hanley, K.A.; Rodriguez, D.N.; et al. PacBio-based mitochondrial genome assembly of Leucaena trichandra (leguminosae) and an intrageneric assessment of mitochondrial RNA editing. Genome Biol. Evol. 2018, 10, 2501–2517. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.; Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef] [Green Version]
- Fajardo, D.; Schlautman, B.; Steffan, S.; Polashock, J.; Vorsa, N.; Zalapa, J. The American cranberry mitochondrial genome reveals the presence of selenocysteine (tRNA-Sec and SECIS) insertion machinery in land plants. Gene 2014, 536, 336–343. [Google Scholar] [CrossRef]
- Rivarola, M.; Foster, J.T.; Chan, A.P.; Williams, A.L.; Rice, D.W.; Liu, X.; Melake-Berhan, A.; Huot Creasy, H.; Puiu, D.; Rosovitz, M.J.; et al. Castor bean organelle genome sequencing and worldwide genetic diversity analysis. PLoS ONE 2011, 6, e21743. [Google Scholar] [CrossRef] [Green Version]
- Magee, A.M.; Aspinall, S.; Rice, D.W.; Cusack, B.P.; Sémon, M.; Perry, A.S.; Stefanović, S.; Milbourne, D.; Barth, S.; Palmer, J.D.; et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010, 20, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goremykin, V.V.; Salamini, F.; Velasco, R.; Viola, R. Mitochondrial DNA of Vitis vinifera and the Issue of Rampant Horizontal Gene Transfer. Mol. Biol. Evol. 2008, 26, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Wang, Y.; Lu, J.; Gai, J.; Li, J.; Chu, P.; Guan, R.; Zhao, T. The mitochondrial genome of soybean reveals complex genome structures and gene evolution at intercellular and phylogenetic levels. PLoS ONE 2013, 8, e56502. [Google Scholar]
- Petersen, G.; Cuenca, A.; Zervas, A.; Ross, G.T.; Graham, S.W.; Barrett, C.F.; Davis, J.I.; Seberg, O. Mitochondrial genome evolution in Alismatales: Size reduction and extensive loss of ribosomal protein genes. PLoS ONE 2017, 12, e177606. [Google Scholar] [CrossRef] [Green Version]
- Clifton, S.W.; Minx, P.; Fauron, C.M.R.; Gibson, M.; Allen, J.O.; Sun, H.; Thompson, M.; Barbazuk, W.B.; Kanuganti, S.; Tayloe, C.; et al. Sequence and Comparative Analysis of the Maize NB Mitochondrial Genome. Plant Physiol. 2004, 136, 3486–3503. [Google Scholar] [CrossRef] [Green Version]
- Cui, P.; Liu, H.; Lin, Q.; Ding, F.; Zhuo, G.; Hu, S.; Liu, D.; Yang, W.; Zhan, K.; Zhang, A.; et al. A complete mitochondrial genome of wheat (Triticum aestivum cv. Chinese Yumai), and fast evolving mitochondrial genes in higher plants. J. Genet. 2009, 88, 299–307. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Watase, Y.; Nagase, M.; Makita, N.; Yagura, S.; Hirai, A.; Sugiura, M. The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: Comparative analysis of mitochondrial genomes in higher plants. Mol. Genet. Genomics 2005, 272, 603–615. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; Mcginnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. Prottest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Odahara, M.; Kuroiwa, H.; Kuroiwa, T.; Sekine, Y. Suppression of repeat-mediated gross mitochondrial genome rearrangements by RecA in the moss physcomitrella patens. Plant Cell. 2009, 21, 1182–1194. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Stone, J.D.; Atorchová, H.; Sloan, D.B. High transcript abundance, RNA editing, and small RNAs in intergenic regions within the massive mitochondrial genome of the angiosperm Silene noctiflora. BMC Genomics. 2015, 16, 938. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Wang, Y.; Hua, J. The roles of mitochondrion in intergenomic gene transfer in plants: A source and a pool. Int. J. Mol. Sci. 2018, 19, 547. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Hu, Y.; He, M.; Zhang, B.; Wu, W.; Cai, P.; Huo, D.; Hong, Y. Comparative chloroplast genomes: Insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia. BMC Genom. 2021, 22, 138. [Google Scholar] [CrossRef]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Law, S.R.; Narsai, R.; Taylor, N.L.; Delannoy, E.; Carrie, C.; Giraud, E.; Millar, A.H.; Small, I.; Whelan, J. Nucleotide and RNA metabolism prime translational initiation in the earliest events of mitochondrial biogenesis during Arabidopsis germination. Plant Physiol. 2012, 158, 1610–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, D.C.; Millar, A.H.; Sweetlove, L.J.; Hill, S.A.; Leaver, C.J. Mitochondrial biogenesis during germination in maize embryos. Plant Physiol. 2001, 125, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Kubo, T.; Nishizawa, S.; Estiati, A.; Itchoda, N.; Mikami, T. The cytoplasmic male-sterile type and normal type mitochondrial genomes of sugar beet share the same complement of genes of known function but differ in the content of expressed ORFs. Mol. Genet. Genomics. 2004, 272, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, D.J.; Bendich, A.J. Mitochondrial DNA from the liverwort marchantia polymorpha: Circularly permuted linear molecules, head-to-tail concatemers, and a 5′ protein 1 1edited by n.-M. Chua. J. Mol. Biol. 2001, 310, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Newton, K.J. Plant mitochondrial genomes: Dynamics and mechanisms of mutation. Annu. Rev. Plant. Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef] [PubMed]
- Cole, L.W.; Guo, W.; Mower, J.P.; Palmer, J.D.; Purugganan, M. High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol. Biol. Evol. 2018, 35, 2773–2785. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, S.; Shi, L.; Gong, D.; Zhang, S.; Zhao, Q.; Zhan, D.; Vasseur, L.; Wang, Y.; Yu, J.; et al. Haplotype-resolved genome assembly provides insights into evolutionary history of the tea plant Camellia sinensis. Nat. Genet. 2021, 53, 1250–1259. [Google Scholar] [CrossRef]
- Wu, Q.; Tong, W.; Zhao, H.; Ge, R.; Li, R.; Huang, J.; Li, F.; Wang, Y.; Mallano, A.I.; Deng, W.; et al. Comparative transcriptomic analysis unveils the deep phylogeny and secondary metabolite evolution of 116 Camellia plants. Plant J. 2022, 111, 406–421. [Google Scholar] [CrossRef]
- Wang, X.; Feng, H.; Chang, Y.; Ma, C.; Wang, L.; Hao, X.; Li, A.L.; Cheng, H.; Wang, L.; Cui, P.; et al. Population sequencing enhances understanding of tea plant evolution. Nat. Commun. 2020, 11, 4447. [Google Scholar] [CrossRef]
- Xia, E.; Tong, W.; Hou, Y.; An, Y.; Chen, L.; Wu, Q.; Liu, Y.; Yu, J.; Li, F.; Li, R.; et al. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mol. Plant. 2020, 13, 1013–1026. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Li, W.; Li, K.; Nan, H.; Shi, C.; Zhang, Y.; Dai, Z.Y.; Lin, Y.L.; Yang, X.L.; Tong, Y.; et al. The chromosome-level reference genome of tea tree unveils recent bursts of non-autonomous LTR retrotransposons in driving genome size evolution. Mol. Plant 2020, 13, 935–938. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Y.; Qiu, H.; Guo, Y.; Wan, H.; Zhang, X.; Scossa, F.; Alseekh, S.; Zhang, Q.; Wang, P.; et al. Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties. Nat. Commun. 2020, 11, 3719. [Google Scholar] [CrossRef]
- Lin, P.; Wang, K.; Wang, Y.; Hu, Z.; Yan, C.; Huang, H.; Ma, X.; Cao, Y.; Long, W.; Liu, W.; et al. The genome of oil-camellia and population genomics analysis provide insights into seed oil domestication. Genome Biol. 2022, 23, 14. [Google Scholar] [CrossRef]
- Sandhya, S.; Srivastava, H.; Kaila, T.; Tyagi, A.; Gaikwad, K. Methods and tools for plant organelle genome sequencing, assembly, and downstream analysis. In Legume Genomics; Humana: New York, NY, USA, 2020; Volume 2107, pp. 49–98. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | C. gigantocarpa | C. sinensis | A. thaliana | |||
|---|---|---|---|---|---|---|
| Length (bp) | Percentage (%) | Length (bp) | Percentage (%) | Length (bp) | Percentage (%) | |
| RNA/non-LTR-RTs | 432 | 0.04 | 992 | 0.11 | 1567 | 0.43 |
| RNA/LTR-RTs | 197,860 | 20.39 | 198,371 | 21.68 | 12,887 | 3.51 |
| DNA transposons | 0 | 0 | 0 | 0 | 104 | 0.03 |
| Other repeats | 3622 | 0.38 | 3315 | 0.36 | 3670 | 0.99 |
| Total | 201,914 | 20.81 | 202,678 | 22.15 | 18,228 | 4.96 |
| SSR Motif | SSR Number | SSR (%) |
|---|---|---|
| Monomer | 242 | 32.44 |
| Dimer | 332 | 44.5 |
| Trimer | 37 | 4.96 |
| Tetramer | 106 | 14.21 |
| Pentamer | 23 | 3.08 |
| Hexamer | 6 | 0.8 |
| Total | 746 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.; Gao, L.-Z.; Zhang, Q.-J. A High-Quality Genome Assembly of the Mitochondrial Genome of the Oil-Tea Tree Camellia gigantocarpa (Theaceae). Diversity 2022, 14, 850. https://doi.org/10.3390/d14100850
Lu C, Gao L-Z, Zhang Q-J. A High-Quality Genome Assembly of the Mitochondrial Genome of the Oil-Tea Tree Camellia gigantocarpa (Theaceae). Diversity. 2022; 14(10):850. https://doi.org/10.3390/d14100850
Chicago/Turabian StyleLu, Cui, Li-Zhi Gao, and Qun-Jie Zhang. 2022. "A High-Quality Genome Assembly of the Mitochondrial Genome of the Oil-Tea Tree Camellia gigantocarpa (Theaceae)" Diversity 14, no. 10: 850. https://doi.org/10.3390/d14100850
APA StyleLu, C., Gao, L. -Z., & Zhang, Q. -J. (2022). A High-Quality Genome Assembly of the Mitochondrial Genome of the Oil-Tea Tree Camellia gigantocarpa (Theaceae). Diversity, 14(10), 850. https://doi.org/10.3390/d14100850