


Assessing Genetic Diversity and Searching for Selection Signatures by Comparison between the Indigenous Livni and Duroc Breeds in Local Livestock of the Central Region of Russia

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Genotyping
2.2. Quality Control
2.3. Genetic Diversity, PCA, Neighbor-Net and Admixture
2.4. Selection Signature Analysis
2.4.1. FST analysis
2.4.2. Runs of Homozygosity Estimation
2.4.3. HapFLK Analysis
2.5. Identification of Candidate Genes
2.6. Functional Enrichment Analysis
3. Results
3.1. Genetic Diversity, Breed Relationship and Admixture
3.2. Selection Signature Detection
3.3. Candidate Gene Determination
3.4. Functional Enrichment Determination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Elbeltagy, A.R. Sheep Genetic Diversity and Breed Differences for Climate-Change Adaptation. In Sheep Production Adapting to Climate Change, 1st ed.; Sejian, V., Bhatta, R., Gaughan, J., Malik, P., Naqvi, S., Lal, R., Eds.; Springer: Singapore, 2017. [Google Scholar] [CrossRef]
- Boettcher, P.J.; Hoffmann, I.; Baumung, R.; Drucker, A.G.; McManus, C.; Berg, P.; Stella, A.; Nilsen, L.B.; Moran, D.; Naves, M.; et al. Genetic resources and genomics for adaptation of livestock to climate change. Front. Genet. 2015, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsartsianidou, V.; Sánchez-Molano, E.; Kapsona, V.V.; Basdagianni, Z.; Chatziplis, D.; Arsenos, G.; Triantafyllidis, A.; Banos, G. A comprehensive genome-wide scan detects genomic regions related to local adaptation and climate resilience in Mediterranean domestic sheep. Genet. Sel. Evol. 2021, 53, 90. [Google Scholar] [CrossRef]
- Passamonti, M.M.; Somenzi, E.; Barbato, M.; Chillemi, G.; Colli, L.; Joost, S.; Milanesi, M.; Negrini, R.; Santini, M.; Vajana, E.; et al. The Quest for Genes Involved in Adaptation to Climate Change in Ruminant Livestock. Animals 2021, 11, 2833. [Google Scholar] [CrossRef]
- Rauw, W.M.; Rydhmer, L.; Kyriazakis, I.; Øverland, M.; Gilbert, H.; Dekkers, J.C.; Hermesch, S.; Bouquet, A.; Gómez Izquierdo, E.; Louveau, I.; et al. Prospects for sustainability of pig production in relation to climate change and novel feed resources. J. Sci. Food Agric. 2020, 100, 3575–3586. [Google Scholar] [CrossRef] [Green Version]
- Ottosen, M.; Mackenzie, S.G.; Filipe, J.A.N.; Misiura, M.M.; Kyriazakis, I. Changes in the environmental impacts of pig production systems in Great Britain over the last 18 years. Agric. Syst. 2021, 189, 103063. [Google Scholar] [CrossRef]
- Phocas, F.; Belloc, C.; Bidanel, J.; Delaby, L.; Dourmad, J.Y.; Dumont, B.; Ezanno, P.; Fortun-Lamothe, L.; Foucras, G.; Frappat, B.; et al. Review: Towards the agroecological management of ruminants, pigs and poultry through the development of sustainable breeding programmes. II. Breeding strategies. Animal 2016, 10, 1760–1769. [Google Scholar] [CrossRef] [Green Version]
- Gama, L.T.; Martínez, A.M.; Carolino, I.; Landi, V.; Delgado, J.V.; Vicente, A.A.; Vega-Pla, J.L.; Cortés, O.; Sousa, C.O.; BIOPIG Consortium. Genetic structure, relationships and admixture with wild relatives in native pig breeds from Iberia and its islands. Genet. Sel. Evol. 2013, 45, 18. [Google Scholar] [CrossRef] [Green Version]
- Quan, J.; Gao, C.; Cai, Y.; Ge, Q.; Jiao, T.; Zhao, S. Population genetics assessment model reveals priority protection of genetic resources in native pig breeds in China. Glob. Ecol. Conserv. 2020, 21, e00829. [Google Scholar] [CrossRef]
- Herrero-Medrano, J.M.; Megens, H.J.; Groenen, M.A.; Bosse, M.; Pérez-Enciso, M.; Crooijmans, R.P. Whole-genome sequence analysis reveals differences in population management and selection of European low-input pig breeds. BMC Genom. 2014, 15, 601. [Google Scholar] [CrossRef]
- Hlongwane, N.L.; Hadebe, K.; Soma, P.; Dzomba, E.F.; Muchadeyi, F.C. Genome Wide Assessment of Genetic Variation and Population Distinctiveness of the Pig Family in South Africa. Front. Genet. 2020, 11, 344. [Google Scholar] [CrossRef]
- Madsen, M.D.; Madsen, P.; Nielsen, B.; Kristensen, T.N.; Jensen, J.; Shirali, M. Macro-environmental sensitivity for growth rate in Danish Duroc pigs is under genetic control. J. Anim. Sci. 2018, 96, 4967–4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.Q.; Xue, J.; Lian, M.J.; Yang, R.F.; Liu, T.F.; Zeng, Z.Y.; Jiang, A.A.; Jiang, Y.Z.; Zhu, L.; Bai, L.; et al. Inbreeding and genetic diversity in three imported Swine breeds in China using pedigree data. Asian-Australas J. Anim. Sci. 2013, 26, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, G.; Ruan, D.; Zhuang, Z.; Ding, R.; Quan, J.; Wang, S.; Jiang, Y.; Huang, J.; Gu, T.; et al. Runs of Homozygosity Uncover Potential Functional-Altering Mutation Associated with Body Weight and Length in Two Duroc Pig Lines. Front. Vet. Sci. 2022, 9, 832633. [Google Scholar] [CrossRef]
- Pavlova, S.V.; Kozlova, N.A. Plemennaya baza svinovodstva Rossii na nachalo 2021 goda Breeding base of pig breeding in Russia at the beginning of 2021. Ehffektivnoe Zhivotnovodstvo 2021, 5, 28–31. (In Russian) [Google Scholar]
- Martins, T.D.; Lemos, M.V.; Mueller, L.F.; Baldi, F.; Amorim, T.; Ferrinho, A.M.; Muñoz, J.A.; Fuzikawa, I.H.; Moura, G.V.; Gemelli, J.L.; et al. Fat Deposition, Fatty Acid Composition, and Its Relationship with Meat Quality and Human Health. In Meat Science and Nutrition, 1st ed.; Arshad, M.S., Ed.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Poklukar, K.; Čandek-Potokar, M.; Batorek Lukač, N.; Tomažin, U.; Škrlep, M. Lipid Deposition and Metabolism in Local and Modern Pig Breeds: A Review. Animals 2020, 10, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razmaitė, V.; Juška, R.; Leikus, R.; Jatkauskienė, V. Pork Quality of Two Lithuanian Breeds: Effects of Breed, Gender and Feeding Regimen. Animals 2021, 11, 1103. [Google Scholar] [CrossRef] [PubMed]
- Baer, A.A.; Dilger, A.C. Effect of fat quality on sausage processing, texture, and sensory characteristics. Meat Sci. 2014, 96, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Tyapugin, S.E.; Butusov, D.V. Yearbook on Breeding Work in Pig Breeding in the Farms of the Russian Federation in 2020; Tyapugin, S.E., Butusov, D.V., Eds.; The Publishing House of All-Russian Research Institute of Animal Breeding of the Russian Ministry for Agriculture: Moscow, Russia, 2021; p. 154. [Google Scholar]
- Nikulnikov, V.S.; Ovsyannikova, N.N.; Lyakhova, O.L.; Sysoeva, L.A.; Efanov, A.M. Livni breed of pigs-a valuable gene pool of Russia. Curr. Probl. Nat. Sci. Educ. Environ. Prot. Hum. Health 2016, 4, 251–255. [Google Scholar]
- Traspov, A.; Deng, W.; Kostyunina, O.; Ji, J.; Shatokhin, K.; Lugovoy, S.; Zinovieva, N.; Yang, B.; Huang, L. Population structure and genome characterization of local pig breeds in Russia, Belorussia, Kazakhstan and Ukraine. Genet. Sel. Evol. 2016, 48, 16. [Google Scholar] [CrossRef] [Green Version]
- Kharzinova, V.R.; Akopyan, N.A.; Dotsev, A.V.; Deniskova, T.E.; Sermyagin, A.A.; Karpushkina, T.V.; Solovieva, A.D.; Brem, G.; Zinovieva, N.A. Genetic Diversity and Phylogenetic Relationships of Russian Pig Breeds Based on the Analysis of mtDNA D-Loop Polymorphism. Russ. J. Genet. 2022, 58, 944–954. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. diveRsity: An R package for the estimation of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis, 1st ed.; Springer: New York, NY, USA, 2009; p. 212. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Francis, R.M. pophelper: An R package and web app to analyse and visualise population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. International Sheep Genomics Consortium Members. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 2015, 47, 49. [Google Scholar] [CrossRef]
- Biscarini, F.; Paolo Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity And Runs Of Heterozygosity In Diploid Genomes. R Package Version 0.9.5. Available online: https://cran.r-project.org/web/packages/detectRUNS/index.html (accessed on 8 May 2021).
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef] [Green Version]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [Green Version]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [Green Version]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.C.D.C.; Ventura, R.V.; Baldi, F.; da Silva, M.V.G.B. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef] [Green Version]
- Grilz-Seger, G.; Neuditschko, M.; Ricard, A.; Velie, B.; Lindgren, G.; Mesarič, M.; Cotman, M.; Horna, M.; Dobretsberger, M.; Brem, G.; et al. Genome-Wide Homozygosity Patterns and Evidence for Selection in a Set of European and Near Eastern Horse Breeds. Genes 2019, 10, 491. [Google Scholar] [CrossRef] [Green Version]
- Fariello, M.I.; Boitard, S.; Naya, H.; SanCristobal, M.; Servin, B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics 2013, 193, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Scheet, P.; Stephens, M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sherman, B.; Lempicki, R. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- GENE ONTOLOGY. Available online: http://geneontology.org/ (accessed on 8 May 2021).
- Zinovieva, N.A.; Sheiko, I.P.; Dotsev, A.V.; Sheiko, R.I.; Mikhailova, M.E.; Sermyagin, A.A.; Abdelmanova, A.S.; Kharzinova, V.R.; Reyer, H.; Wimmers, K.; et al. Genome-wide SNP analysis clearly distinguished the Belarusian Red cattle from other European cattle breeds. Anim. Genet. 2021, 52, 720–724. [Google Scholar] [CrossRef]
- Abdelmanova, A.S.; Sermyagin, A.A.; Dotsev, A.V.; Rodionov, A.N.; Stolpovsky, Y.A.; Zinovieva, N.A. Whole-Genomic Studies of the Population Structure of Russian Local Black-Pied Breeds. Russ. J. Genet. 2022, 58, 804–813. [Google Scholar] [CrossRef]
- Lukić, B.; Ferenčaković, M.; Šalamon, D.; Čačić, M.; Orehovački, V.; Iacolina, L.; Curik, I.; Cubric-Curik, V. Conservation Genomic Analysis of the Croatian Indigenous Black Slavonian and Turopolje Pig Breeds. Front. Genet. 2020, 11, 261. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Bertolini, F.; Tinarelli, S.; Dall’Olio, S.; Nanni Costa, L.; Gallo, M.; Fontanesi, L. Comparative evaluation of genomic inbreeding parameters in seven commercial and autochthonous pig breeds. Animal 2020, 14, 910–920. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, K.; Peng, X.; Zhan, H.; Lu, J.; Xie, S.; Zhao, S.; Li, X.; Ma, Y. Selective sweep analysis reveals extensive parallel selection traits between large white and Duroc pigs. Evol. Appl. 2020, 13, 2807–2820. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Zhang, B.; Zhong, H.; Lu, Y.; Zhang, H. Candidate gene screening for lipid deposition using combined transcriptomic and proteomic data from Nanyang black pigs. BMC Genom. 2021, 22, 441. [Google Scholar] [CrossRef] [PubMed]
- Xing, K.; Wang, K.; Ao, H.; Chen, S.; Tan, Z.; Wang, Y.; Xitong, Z.; Yang, T.; Zhang, F.; Liu, Y.; et al. Comparative adipose transcriptome analysis digs out genes related to fat deposition in two pig breeds. Sci. Rep. 2019, 9, 12925. [Google Scholar] [CrossRef] [Green Version]
- Zambonelli, P.; Gaffo, E.; Zappaterra, M.; Bortoluzzi, S.; Davoli, R. Transcriptional profiling of subcutaneous adipose tissue in Italian Large White pigs divergent for backfat thickness. Anim. Genet. 2016, 47, 306–323. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Ballester, M.; Fortes, M.R.; Esteve-Codina, A.; Castelló, A.; Noguera, J.L.; Fernández, A.I.; Pérez-Enciso, M.; Reverter, A.; Folch, J.M. From SNP co-association to RNA co-expression: Novel insights into gene networks for intramuscular fatty acid composition in porcine. BMC Genom. 2014, 15, 232. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Wu, P.; Yang, Q.; Wang, K.; Zhou, J.; Yang, X.; Jiang, A.; Shen, L.; Xiao, W.; Jiang, Y.; et al. Genome-wide association study for backfat thickness at 100 kg and loin muscle thickness in domestic pigs based on genotyping by sequencing. Physiol. Genom. 2019, 51, 261–266. [Google Scholar] [CrossRef]
- Yang, B.; Bassols, A.; Saco, Y.; Pérez-Enciso, M. Association between plasma metabolites and gene expression profiles in five porcine endocrine tissues. Genet. Sel. Evol. 2011, 43, 28. [Google Scholar] [CrossRef]
- Xing, K.; Zhao, X.; Ao, H.; Chen, S.; Yang, T.; Tan, Z.; Wang, Y.; Zhang, F.; Liu, Y.; Ni, H.; et al. Transcriptome analysis of miRNA and mRNA in the livers of pigs with highly diverged backfat thickness. Sci. Rep. 2019, 9, 16740. [Google Scholar] [CrossRef] [Green Version]
- Wucher, V.; Sodaei, R.; Amador, R.; Irimia, M.; Guigó, R. Day-night and seasonal variation of human gene expression across tissues. bioRxiv 2022, 433266, preprint. [Google Scholar] [CrossRef]
- Srikanth, K.; Lee, S.-H.; Chung, K.-Y.; Park, J.-E.; Jang, G.-W.; Park, M.-R.; Kim, N.Y.; Kim, T.-H.; Chai, H.-H.; Park, W.C.; et al. A Gene-Set Enrichment and Protein–Protein Interaction Network-Based GWAS with Regulatory SNPs Identifies Candidate Genes and Pathways Associated with Carcass Traits in Hanwoo Cattle. Genes 2020, 11, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguita-Ruiz, A.; Bustos-Aibar, M.; Plaza-Díaz, J.; Mendez-Gutierrez, A.; Alcalá-Fdez, J.; Aguilera, C.M.; Ruiz-Ojeda, F.J. Omics Approaches in Adipose Tissue and Skeletal Muscle Addressing the Role of Extracellular Matrix in Obesity and Metabolic Dysfunction. Int. J. Mol. Sci. 2021, 22, 2756. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, M.; Liang, C.; Sheng, N.; Wang, X.; Wang, F.; Luo, Y.; Jiang, J.; Cai, L.; Niu, H.; et al. Anterior gradient 2 increases long-chain fatty acid uptake via stabilizing FABP1 and facilitates lipid accumulation. Int. J. Biol. Sci. 2021, 17, 834–847. [Google Scholar] [CrossRef]
- Dervishi, E.; González-Calvo, L.; Blanco, M.; Joy, M.; Sarto, P.; Martin-Hernandez, R.; Ordovás, J.M.; Serrano, M.; Calvo, J.H. Gene Expression and Fatty Acid Profiling in Longissimus thoracis Muscle, Subcutaneous Fat, and Liver of Light Lambs in Response to Concentrate or Alfalfa Grazing. Front. Genet. 2019, 10, 1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romao, J.M.; Jin, W.; He, M.; McAllister, T.; Guan, L.L. Elucidation of Molecular Mechanisms of Physiological Variations between Bovine Subcutaneous and Visceral Fat Depots under Different Nutritional Regimes. PLoS ONE 2013, 8, e83211. [Google Scholar] [CrossRef]
- Pan, C.; Lei, Z.; Wang, S.; Wang, X.; Wei, D.; Cai, X.; Luoreng, Z.; Wang, L.; Ma, Y. Genome-wide identification of cyclin-dependent kinase (CDK) genes affecting adipocyte differentiation in cattle. BMC Genom. 2021, 22, 532. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, R.N.; Zachara, M.; Deplancke, B. A systems perspective on brown adipogenesis and metabolic activation. Obes. Rev. 2017, 18, 65–81. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Hill, R.C.; Dzieciatkowska, M.; Zhu, L.; Infante, A.M.; Hu, G.; Hansen, K.C.; Pei, M. Site-Dependent Lineage Preference of Adipose Stem Cells. Front. Cell Dev. Biol. 2020, 8, 237. [Google Scholar] [CrossRef]
- Massiera, F.; Barbry, P.; Guesnet, P.; Joly, A.; Luquet, S.; Moreilhon-Brest, C.; Mohsen-Kanson, T.; Amri, E.Z.; Ailhaud, G. A Western-like fat diet is sufficient to induce a gradual enhancement in fat mass over generations. J. Lipid Res. 2010, 51, 2352–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Xie, S.; Jin, W. Crucial lncRNAs associated with adipocyte differentiation from human adipose-derived stem cells based on co-expression and ceRNA network analyses. PeerJ 2019, 7, e7544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, P.A.; Wahlstrand, B.; Olsson, M.; Froguel, P.; Falchi, M.; Bergman, R.N.; McTernan, P.G.; Hedner, T.; Carlsson, L.M.; Jacobson, P. CDKN2B expression and subcutaneous adipose tissue expandability: Possible influence of the 9p21 atherosclerosis locus. Biochem. Biophys. Res. Commun. 2014, 446, 1126–1131. [Google Scholar] [CrossRef] [Green Version]
- Wing, M.R. The Genetics of Differential Fat Distribution: The Insulin Resistance Atherosclerosis Family Study. For the Degree of Doctor of Philosophy in the Molecular Genetics and Genomics Program; Wake Forest University Graduate School of Arts And Sciences: Winston-Salem, NC, USA, 2010. [Google Scholar]
- Zappaterra, M.; Gioiosa, S.; Chillemi, G.; Zambonelli, P.; Davoli, R. Muscle transcriptome analysis identifies genes involved in ciliogenesis and the molecular cascade associated with intramuscular fat content in Large White heavy pigs. PLoS ONE 2020, 15, e0233372. [Google Scholar] [CrossRef]
- Sun, C.; Kovacs, P.; Guiu-Jurado, E. Genetics of Body Fat Distribution: Comparative Analyses in Populations with European, Asian and African Ancestries. Genes 2021, 12, 841. [Google Scholar] [CrossRef]
- Mačeková, S.; Mathia, M.; Varaliová, Z.; Bernasovská, J.; Boroňová, Y.; Dojčaková, D.; Petrejčíková, E.; Galová, J.; Vašková, H. Ethnic difference frequencies of THSD7A variant, but no association with obesity. Anthropologie 2021, 59, 205–211. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, C.; Sun, Y.; Li, Y.; Kang, L.; Jiang, Y. Dynamic transcriptome and DNA methylome analyses on longissimus dorsi to identify genes underlying intramuscular fat content in pigs. BMC Genom. 2017, 18, 780. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Huang, Z.; Zhao, W.; Li, M.; Li, C. Transcriptome Analysis Reveals Long Intergenic Non-Coding RNAs Contributed to Intramuscular Fat Content Differences between Yorkshire and Wei Pigs. Int. J. Mol. Sci. 2020, 21, 1732. [Google Scholar] [CrossRef] [Green Version]
- Raschetti, M.; Rignanese, D.; Pagnacco, G.; Castiglioni, B.; Chessa, S. Polymorphisms in swine candidate genes for meat quality detected by PCR-SSCP. Ital. J. Anim. Sci. 2009, 8, 129–131. [Google Scholar] [CrossRef]
- Tao, X.; Liang, Y.; Yang, X.; Pang, J.; Zhong, Z.; Chen, X.; Yang, Y.; Zeng, K.; Kang, R.; Lei, Y.; et al. Transcriptomic profiling in muscle and adipose tissue identifies genes related to growth and lipid deposition. PLoS ONE 2017, 12, e0184120. [Google Scholar] [CrossRef]
- Gottmann, P.; Ouni, M.; Saussenthaler, S.; Roos, J.; Stirm, L.; Jähnert, M.; Kamitz, A.; Hallahan, N.; Jonas, W.; Fritsche, A.; et al. A computational biology approach of a genome-wide screen connected miRNAs to obesity and type 2 diabetes. Mol. Metab. 2018, 11, 145–159. [Google Scholar] [CrossRef]
- Sanchez, E.M. Modulation of Porcine Production And Molecular Phenotypes By Nutrition And Genetics. Ph.D. Thesis, Universitat Autonoma de Barcelona, Barcelona, Spain, June 2020. [Google Scholar]
- Gupta, V.A.; Beggs, A.H. Kelch proteins: Emerging roles in skeletal muscle development and diseases. Skelet. Muscle 2014, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Li, N.; Cheng, H.; Ma, Y. Genome wide association study for the identification of genes associated with tail fat deposition in Chinese sheep breeds. Biol. Open 2021, 10, bio054932. [Google Scholar] [CrossRef]
- Fan, B.; Onteru, S.K.; Mote, B.E.; Serenius, T.; Stalder, K.J.; Rothschild, M.F. Large-scale association study for structural soundness and leg locomotion traits in the pig. Genet. Sel. Evol. 2009, 41, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Xiao, Q.; Zhang, Q.; Sun, H.; Chen, J.; Li, Z.; Xue, M.; Ma, P.; Yang, H.; Xu, N.; et al. Genomic analysis reveals genes affecting distinct phenotypes among different Chinese and western pig breeds. Sci. Rep. 2018, 8, 13352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlakõiv, T.; Flamar, A.L.; Johnston, L.K.; Moriyama, S.; Putzel, G.G.; Bryce, P.J.; Artis, D. Stromal cells maintain immune cell homeostasis in adipose tissue via production of interleukin-33. Sci. Immunol. 2019, 4, eaax0416. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Lee, M.W.; Sogawa, Y.; Bertholet, A.M.; Locksley, R.M.; Weinberg, D.E.; Kirichok, Y.; Deo, R.C.; Chawla, A. Perinatal Licensing of Thermogenesis by IL-33 and ST2. Cell 2016, 166, 841–854. [Google Scholar] [CrossRef] [Green Version]
- Fyda, T.J.; Spencer, C.; Jastroch, M.; Gaudry, M.J. Disruption of thermogenic UCP1 predated the divergence of pigs and peccaries. J. Exp. Biol. 2020, 223, jeb223974. [Google Scholar] [CrossRef]
- Jastroch, M.; Andersson, L. When pigs fly, UCP1 makes heat. Mol. Metab. 2015, 4, 359–362. [Google Scholar] [CrossRef]
- Zhao, J.; Tao, C.; Chen, C.; Wang, Y.; Liu, T. Formation of thermogenic adipocytes: What we have learned from pigs. Fundam. Res. 2021, 1, 495–502. [Google Scholar] [CrossRef]
- Lin, J.; Cao, C.; Tao, C.; Ye, R.; Dong, M.; Zheng, Q.; Wang, C.; Jiang, X.; Qin, G.; Yan, C.; et al. Cold adaptation in pigs depends on UCP3 in beige adipocytes. J. Mol. Cell Biol. 2017, 9, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Zhang, Y.; Li, W.; Hu, A.J.; Luo, C.; Zhou, W.; Hu, J.K.; Daniele, S.G.; Wang, J.; Sheng, J.; et al. CDK6 inhibits white to beige fat transition by suppressing RUNX1. Nat. Commun. 2018, 9, 1023. [Google Scholar] [CrossRef] [Green Version]
- Duta-Mare, M.; Sachdev, V.; Leopold, C.; Kolb, D.; Vujic, N.; Korbelius, M.; Hofer, D.C.; Xia, W.; Huber, K.; Auer, M.; et al. Lysosomal acid lipase regulates fatty acid channeling in brown adipose tissue to maintain thermogenesis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, S.; Hallen, S.; Huang, L.; Svensson, P.A.; Momo, R.A.; Wallin, S.; Carlsson, E.K.; Forslöw, A.; Seale, P.; Peng, X.R. Thermogenic activity of UCP1 in human white fat-derived beige adipocytes. Mol. Endocrinol. 2015, 29, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Kahoul, Y.; Oger, F.; Montaigne, J.; Froguel, P.; Breton, C.; Annicotte, J.-S. Emerging Roles for the INK4a/ARF (CDKN2A) Locus in Adipose Tissue: Implications for Obesity and Type 2 Diabetes. Biomolecules 2020, 10, 1350. [Google Scholar] [CrossRef]
- Milet, C.; Bléher, M.; Allbright, K.; Orgeur, M.; Coulpier, F.; Duprez, D.; Havis, E. Egr1 deficiency induces browning of inguinal subcutaneous white adipose tissue in mice. Sci. Rep. 2017, 7, 16153. [Google Scholar] [CrossRef] [Green Version]
- Reynés, B.; van Schothorst, E.M.; Keijer, J.; Ceresi, E.; Oliver, P.; Palou, A. Cold Induced Depot-Specific Browning in Ferret Aortic Perivascular Adipose Tissue. Front. Physiol. 2019, 10, 1171. [Google Scholar] [CrossRef] [Green Version]
- Kiskinis, E.; Chatzeli, L.; Curry, E.; Kaforou, M.; Frontini, A.; Cinti, S.; Montana, G.; Parker, M.G.; Christian, M. RIP140 represses the “brown-in-white” adipocyte program including a futile cycle of triacylglycerol breakdown and synthesis. Mol. Endocrinol. 2014, 28, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, A.A.; Deniskova, T.E.; Yudin, N.S.; Dotsev, A.V.; Khamiruev, T.N.; Selionova, M.I.; Egorov, S.V.; Reyer, H.; Wimmers, K.; Brem, G.; et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC Genom. 2019, 20, 294. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.H.; Agha, S.; Kantanen, J.; Colli, L.; Stucki, S.; Kijas, J.W.; Joost, S.; Li, M.H.; Ajmone Marsan, P. Adaptations to climate-mediated selective pressures in sheep. Mol. Biol. Evol. 2014, 31, 3324–3343. [Google Scholar] [CrossRef]
- Kim, E.S.; Elbeltagy, A.R.; Aboul-Naga, A.M.; Rischkowsky, B.; Sayre, B.; Mwacharo, J.M.; Rothschild, M.F. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity 2016, 116, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Yudin, N.S.; Larkin, D.M.; Ignatieva, E.V. A compendium and functional characterization of mammalian genes involved in adaptation to Arctic or Antarctic environments. BMC Genet. 2017, 18, 111. [Google Scholar] [CrossRef] [Green Version]
- Freitas, P.H.F.; Wang, Y.; Yan, P.; Oliveira, H.R.; Schenkel, F.S.; Zhang, Y.; Xu, Q.; Brito, L.F. Genetic Diversity and Signatures of Selection for Thermal Stress in Cattle and Other Two Bos Species Adapted to Divergent Climatic Conditions. Front. Genet. 2021, 12, 604823. [Google Scholar] [CrossRef]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting Group. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Glaser, J.; Iranzo, J.; Borensztein, M.; Marinucci, M.; Gualtieri, A.; Jouhanneau, C.; Teissandier, A.; Gaston-Massuet, C.; Bourc’his, D. The imprinted Zdbf2 gene finely tunes control of feeding and growth in neonates. Elife 2022, 11, e65641. [Google Scholar] [CrossRef] [PubMed]
- Kominakis, A.; Tarsani, E.; HagerTheodorides, A.L.; Mastranestasis, I.; Gkelia, D.; Hadjigeorgiou, I. Genetic differentiation of mainland-island sheep of Greece: Implications for identifying candidate genes for long-term local adaptation. PLoS ONE 2021, 16, e0257461. [Google Scholar] [CrossRef] [PubMed]
- Pant, R.; Firmal, P.; Shah, V.K.; Alam, A.; Chattopadhyay, S. Epigenetic Regulation of Adipogenesis in Development of Metabolic Syndrome. Front. Cell. Dev. Biol. 2021, 8, 619888. [Google Scholar] [CrossRef]
- Alshargabi, R.; Shinjo, T.; Iwashita, M.; Yamashita, A.; Sano, T.; Nishimura, Y.; Hayashi, M.; Zeze, T.; Fukuda, T.; Sanui, T.; et al. SPOCK1 induces adipose tissue maturation: New insights into the function of SPOCK1 in metabolism. Biochem. Biophys. Res. Commun. 2020, 533, 1076–1082. [Google Scholar] [CrossRef]
- Richard, A.J.; Stephens, J.M. The role of JAK-STAT signaling in adipose tissue function. Biochim. Biophys. Acta 2014, 1842, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Cottam, M.A.; Caslin, H.L.; Winn, N.C.; Hasty, A.H. Multiomics reveals persistence of obesity-associated immune cell phenotypes in adipose tissue during weight loss and subsequent weight regain. bioRxiv 2021, 455954, preprint. [Google Scholar] [CrossRef]
- Tripathi, S.; Pohl, M.O.; Zhou, Y.; Rodriguez-Frandsen, A.; Wang, G.; Stein, D.A.; Moulton, H.M.; DeJesus, P.; Che, J.; Mulder, L.C.; et al. Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 2015, 18, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Avilés, C.; Polvillo, O.; Peña, F.; Juárez, M.; Martínez, A.L.; Molina, A. Associations between DGAT1, FABP4, LEP, RORC, and SCD1 gene polymorphisms and fat deposition in Spanish commercial beef. J. Anim. Sci. 2013, 91, 4571–4577. [Google Scholar] [CrossRef]
- Mesas, L.C. Functional Analysis of Candidate Genes For Meat Quality Traits And Muscle Transcriptomics In Pigs. Ph.D. Thesis, Universitat Autonoma de Barcelona, Barcelona, Spain, 2020. [Google Scholar]
- Kim, S.; Lee, Y.M.; Kim, D.H.; Ha, J.J.; Yi, J.K.; Kim, D.H.; Oh, D.; Han, K. Investigation of high correlation with carcass traits of SNPs of the PLCB1, C/EBPα, and TDRKH genes and the combinations of SNPs using the MDR method in the Hanwoo. Genes Genom. 2021, 43, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Taye, M.; Kim, J.; Yoon, S.H.; Lee, W.; Hanotte, O.; Dessie, T.; Kemp, S.; Mwai, O.A.; Caetano-Anolles, K.; Cho, S.; et al. Whole genome scan reveals the genetic signature of African Ankole cattle breed and potential for higher quality beef. BMC Genet. 2017, 18, 11. [Google Scholar] [CrossRef]
- Villanueva, C.J.; Vergnes, L.; Wang, J.; Drew, B.G.; Hong, C.; Tu, Y.; Hu, Y.; Peng, X.; Xu, F.; Saez, E.; et al. Adipose subtype-selective recruitment of TLE3 or Prdm16 by PPARγ specifies lipid storage versus thermogenic gene programs. Cell Metab. 2013, 17, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Gil, S.Y.; Youn, B.S.; Byun, K.; Huang, H.; Namkoong, C.; Jang, P.G.; Lee, J.Y.; Jo, Y.H.; Kang, G.M.; Kim, H.K.; et al. Clusterin and LRP2 are critical components of the hypothalamic feeding regulatory pathway. Nat. Commun. 2013, 4, 1862. [Google Scholar] [CrossRef] [Green Version]
- Fitzgibbons, T.P.; Kogan, S.; Aouadi, M.; Hendricks, G.M.; Straubhaar, J.; Czech, M.P. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1425–H1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morera, J.R. Transcriptomic Analysis Of White And Brown Adipose Tissue During Non-Shivering Termogenesis. Ph.D. Thesis, Universitat Autonoma de Barcelona, Barcelona, Spain, 2019. [Google Scholar]
- Ullah, M.; Stich, S.; Häupl, T.; Eucker, J.; Sittinger, M.; Ringe, J. Reverse differentiation as a gene filtering tool in genome expression profiling of adipogenesis for fat marker gene selection and their analysis. PLoS ONE 2013, 8, e69754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Yang, J.; He, J.; Gong, Y.; Xiao, Y.; Zeng, Q.; Xu, K.; Duan, Y.; He, J.; Ma, H. Spatiotemporal Regulation and Functional Analysis of Circular RNAs in Skeletal Muscle and Subcutaneous Fat during Pig Growth. Biology 2021, 10, 841. [Google Scholar] [CrossRef] [PubMed]
- Lentini, J.M.; Bargabos, R.; Chen, C.; Fu, D. METTL8 is required for 3-methylcytosine modification in human mitochondrial tRNAs. bioRxiv 2021, 442361, preprint. [Google Scholar] [CrossRef]
- Tobi, E.W.; Slieker, R.C.; Luijk, R.; Dekkers, K.F.; Stein, A.D.; Xu, K.M.; Biobank-based Integrative Omics Studies Consortium; Slagboom, P.E.; van Zwet, E.W.; Lumey, L.H.; et al. DNA methylation as a mediator of the association between prenatal adversity and risk factors for metabolic disease in adulthood. Sci. Adv. 2018, 4, eaao4364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daza, K.R.; Velez-Irizarry, D.; Casiró, S.; Steibel, J.P.; Raney, N.E.; Bates, R.O.; Ernst, C.W. Integrated Genome-Wide Analysis of MicroRNA Expression Quantitative Trait Loci in Pig Longissimus Dorsi Muscle. Front. Genet. 2021, 12, 644091. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.; Lu, Z.H.; Megens, H.J.; Archibald, A.L.; Haley, C.; Jackson, I.J.; Groenen, M.A.; Crooijmans, R.P.; Ogden, R.; Wiener, P. Signatures of diversifying selection in European pig breeds. PLoS Genet. 2013, 9, e1003453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, S.; Hosoda, M.; Yoshino, K.-i.; Yamanoue, M.; Shirai, Y. Gene Expression Analysis Provides New Insights into the Mechanism of Intramuscular Fat Formation in Japanese Black Cattle. Genes 2021, 12, 1107. [Google Scholar] [CrossRef] [PubMed]
- Le, V.; Rohmer, T.; David, I. Impact of environmental disturbances on estimated genetic parameters and breeding values for growth traits in pigs. Animal 2022, 16, 100496. [Google Scholar] [CrossRef]
- Van der Werf, J.H.J. Sustainable animal genetic improvement. E3S Web Conf. 2022, 335, 00001. [Google Scholar] [CrossRef]
- Herrero-Medrano, J.M. Conservation Genetics of Local and Wild Pig Populations: Insight in Genetic Diversity and Demographic History. Ph.D. Thesis, Wageningen University, Wageningen, The Netherlands, 2013. [Google Scholar]



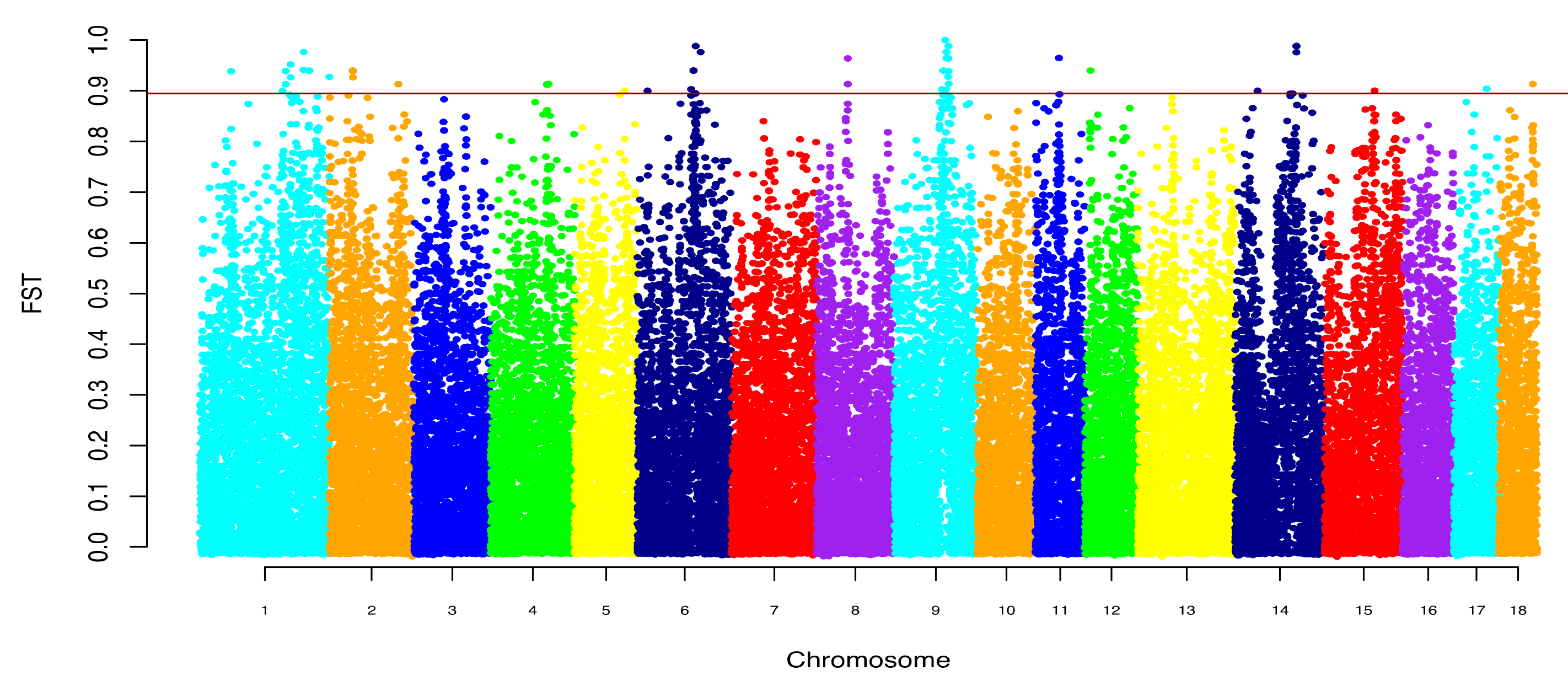{kind=link}
{kind=link}
| SSA * | Breed | Position of Region | Amount of SNP in Region | Length, Mb | The Most Significant SNP | p-Value | |
|---|---|---|---|---|---|---|---|
| Start | End | ||||||
| 1 | DU | 216,980,027 | 244,920,837 | 255 | 27.94 | 231,073,909 | 0.000010 |
| 3 | DU | 45,778,105 | 56,709,945 | 154 | 10.93 | 52,588,363 | 0.000187 |
| 4 | DU, LV | 106,411,407 | 106,750,789 | 10 | 0.34 | 106,485,236 | 0.007915 |
| 6 | DU, LV | 91,706,615 | 101,474,614 | 134 | 9.77 | 97,214,894 | 0.000275 |
| 9 | DU | 82,692,233 | 105,612,360 | 273 | 22.92 | 93,125,776 | 0.000487 |
| 10 | DU | 49,911,155 | 51,759,133 | 61 | 1.85 | 50,699,204 | 0.001653 |
| 12 | DU | 33,122,614 | 33,348,283 | 7 | 0.23 | 33,220,015 | 0.008654 |
| 13 | LV | 65,209,239 | 71,634,446 | 68 | 6.43 | 68,207,174 | 0.001558 |
| 14 | DU | 103,168,211 | 108,016,508 | 89 | 4.85 | 104,929,965 | 0.003601 |
| 15 | LV | 84,563,546 | 88,484,722 | 81 | 3.92 | 85,635,190 | 0.003762 |
| 18 | DU | 53,956,913 | 54,852,652 | 23 | 0.90 | 54,392,049 | 0.006769 |
| SSA * | FSTa | ROHb | hapFLKc | |||
|---|---|---|---|---|---|---|
| Breed | Position | Breed | Position | Breed | Position | |
| 1 | DU/LV | 198,346,039 | DU | 198.0–202.1 | ||
| 1 | DU/LV | 229,476,564; 229,502,611 | DU | 228.6–231.0 | DU | 217.0–244.9 |
| 1 | DU/LV | 272,689,388; 272,760,898 | DU | 271.9–275.6 | ||
| 2 | DU/LV | 31,566,031; 32,186,193; 32,313,049; 32,319,002; 32,407,451 | DU | 31.3–33.6 | ||
| 3 | DU | 49.2–54.7 | DU | 45.7–56.7 | ||
| 4 | DU/LV | 106,698,421; 106,719,032 106,750,789 | DU, LV | 106.4–106.7 | ||
| 6 | DU/LV | 90,705,621; 91,279,252 94,442,844; 94,451,345; 94,775,420; 95,482,175 | DU | 88.0–93.2 93.5–99.2 | DU, LV | 91.7–101.5 |
| 9 | DU/LV | 82,963,397; 85,926,552; 93,596,926; 95,858,320; 97,264,389; 97,527,550; 102,510,717; 103,035,428; 103,174,861; 103,267,375; 104,996,366 | DU | 75.8–106.5 | DU | 82.7–105.6 |
| 10 | DU | 49.5–49.9 | DU | 49.9–51.8 | ||
| 14 | DU/LV | 100,350,445; 104,208,040; 104,282,405; 105,893,370; 106,938,671 | LV | 100.1–101.4 | ||
| DU | 100.1–109.3 | DU | 103.1–108.0 | |||
| 14 | DU/LV | 114,848,572; 114,895,388; 114,958,111 | DU | 110.0–117.8 | ||
| 15 | LV/LW | 84,696,087 | LV | 84.7–85.8 | LV | 84.6–88.5 |
| 15 | LV/LN | 90,388,740 | LV | 90.4–91.4 | ||
| 15 | DU/LV | 121,814,208 | DU | 118.8–121.7 | ||
| 18 | DU/LV | 54,277,674 | DU | 54.1–55.8 | DU | 53.9–54.9 |
| SSA * | The LV Breed | The DU Breed | ||
|---|---|---|---|---|
| Method | Position | Method | Position | |
| 1 | ROH | 71,814,075–72,721,133 | ROH | 71,950,726–72,721,133 |
| 1 | ROH | 83,260,076–84,223,593 | ROH | 83,260,076–84,223,593 |
| 1 | ROH | 241,903,331–242,955,813 | hapFLK | 216,980,027–244,920,837 |
| 6 | ROH | 71,436,086–72,057,699 | ROH | 71,303,189–72,477,552 |
| 11 | ROH | 34,824 047–39,790,178 | ROH | 36,953,937–40,366,928 |
| 14 | ROH | 100,097,831—101,350,552 | ROH | 100,162,325—109,285,369 |
| SSA * | Methods a | Region (Mb) | Genes b |
|---|---|---|---|
| The LV breed | |||
| 15 | ROH, FST, hapFLK | 84.7–85.8 | ABCB11, LRP2, DHRS9, FASTKD1, CCDC173, KLHL23, UBR3, MYO3B, GAD1, GORASP2, TLK1, METTL8, DCAF17, CYBRD1, SLC25A12, METAP1D, DLX1, ITGA6, PDK1, RAPGEF4 |
| 15 | ROH, FST | 90.4–91.4 | LNPK, HOXD13 |
| The DU breed | |||
| 1 | ROH, FST | 198.0–202.1 | LRR1, KLHDC2, SOS2, CDKL1, MAP4K5, ATL1, SAV1, NIN, PYGL, TRIM9, TMX1, FRMD6, RTRAF, NID2 |
| 1 | ROH, FST, hapFLK | 217.0–244.9 | KCNH5, TEK, U6, ELAVL2, ZEB2, CDKN2B, P14ARF, KLHL9, FOCAD, MLLT3, SLC24A2, SAXO1, ADAMTSL1, SH3GL2, CNTLN, BNC2, CCDC171, PSIP1, TTC39B, FREM1, NFIB, MPDZ, LURAP1L, PTPRD, KDM4C, GLDC, RANBP6, IL33, KIAA2026, MLANA, ERMP1, PDCD1LG2, CD274, JAK2, RCL1, SPATA6L, GLIS3, PUM3, KCNV2 |
| 1 | ROH, FST | 271.9–275.6 | GRIN3A, SMC2, OR13C3, ABCA1, NIPSNAP3A |
| 2 | ROH, FST | 31.3–33.6 | PAX6, ELP4, IMMP1L, DCDC1, MPPED2 |
| 3 | ROH, hapFLK | 45.7–56.7 | ZC3H8, MERTK, ACOXL, BUB1, SEPTIN10, NPHP1, ZNF514, PROM2, KCNIP3, ARID5A, NCAPH, SNRNP200, STARD7, SH3RF3, EDAR, CCDC138, RANBP2, SLC5A7, ST6GAL2, UXS1, NCK2, FHL2, GPR45, TGFBRAP1, SLC9A2, IL18RAP, IL18R1, IL1RL1, IL1R1, IL1R2, RFX8, CREG2, RNF149, CNOT11, TBC1D8, CHST10 |
| 9 | ROH, FST, hapFLK | 75.8–106.5 | ZNF804B, CDK14, AKAP9, CYP51A1, ANKIB1, PEX1, CDK6, SAMD9, VPS50, CALCR, GNGT1, COL1A2, CASD1, SGCE, PPP1R9A, PON3, PON2, ASB4, PDK4, U6, DYNC1I1, SLC25A13, TAC1, ASNS, COL28A1, MIOS, UMAD1, GLCCI1, ICA1, NXPH1, CHRDL2, PHF14, THSD7A, TMEM106B, SCIN, DGKB, AGMO, MEOX2, CRPPA, TSPAN13, AGR2, AHR, SNX13, HDAC9, TMEM196, ITGB8, ABCB5, SP4, DNAH11, CDCA7L, RAPGEF5, TOMM7, KLHL7, NUP42, IGF2BP3, RUNDC3B, CROT, ELAPOR2, GRM3, SEMA3A |
| 10 | ROH, hapFLK | 49.5–51.8 | RSU1, C1QL3, PTER, MINDY3, ITGA8, FAM171A1, NMT2, ACBD7, SUV39H2, HSPA14 |
| 14 | ROH, FST, hapFLK | 101.4–109.3 | PRKG1, A1CF |
| 14 | ROH, FST | 110.0–117.8 | CH25H, LIPA, IFIT2, SLC16A12, PANK1, KIF20B, RPP30, PCGF5, HECTD2, BTAF1, CPEB3, MARCHF5, IDE, HHEX, EXOC6, CYP26C1, MYOF, CEP55, PDE6C, FRA10AC1, LGI1, PLCE1, NOC3L, TBC1D12, HELLS, CYP2C42, PDLIM1, SORBS1, ALDH18A1, TCTN3, ENTPD1, CC2D2B, ZNF518A, BLNK, DNTT, OPALIN, TLL2, PIK3AP1 |
| 15 | ROH, FST | 118.8–121.7 | PARD3B, NRP2, INO80D, NDUFS1, ZDBF2, ADAM23, DYTN, CPO |
| 18 | ROH, FST, hapFLK | 53.9–55.8 | CCDC201, ADCY1, CCM2, PURB, MYO1G, ZMIZ2, OGDH, DDX56, NUDCD3, GCK, CAMK2B |
| The LV and the DU breeds | |||
| 1 | ROH | 71.8–72.7 | UFL1, FHL5, GPR63, KLHL32, MMS22L |
| 1 | ROH | 83.2–84.2 | SEC63, OSTM1, SNX3, AFG1L |
| 1 | ROH, hapFLK | 241.9–243.0 | KIAA2026, MLANA, ERMP1, U6, PDCD1LG2, CD274, JAK2 |
| 4 | FST, hapFLK | 106.4–106.7 | RORC, TDRKH, MRPL9, TUFT1, SNX27 |
| 6 | ROH | 71.3–72.5 | ALDH4A1, UBR4, CAPZB |
| 6 | FST, hapFLK | 91.7–101.5 | RAB31, ANKRD12, MTCL1, PTPRM, ARHGAP28, EPB41L3, MYOM1, SMCHD1, NDC80, METTL4, GREB1L, CABLES1, TMEM241, NPC1, LAMA3 |
| 11 | ROH | 34.8–40.4 | TDRD3, SPOCK1 |
| 14 | ROH, FST, hapFLK | 100.1–101.4 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernukha, I.; Abdelmanova, A.; Kotenkova, E.; Kharzinova, V.; Zinovieva, N.A. Assessing Genetic Diversity and Searching for Selection Signatures by Comparison between the Indigenous Livni and Duroc Breeds in Local Livestock of the Central Region of Russia. Diversity 2022, 14, 859. https://doi.org/10.3390/d14100859
Chernukha I, Abdelmanova A, Kotenkova E, Kharzinova V, Zinovieva NA. Assessing Genetic Diversity and Searching for Selection Signatures by Comparison between the Indigenous Livni and Duroc Breeds in Local Livestock of the Central Region of Russia. Diversity. 2022; 14(10):859. https://doi.org/10.3390/d14100859
Chicago/Turabian StyleChernukha, Irina, Alexandra Abdelmanova, Elena Kotenkova, Veronika Kharzinova, and Natalia A. Zinovieva. 2022. "Assessing Genetic Diversity and Searching for Selection Signatures by Comparison between the Indigenous Livni and Duroc Breeds in Local Livestock of the Central Region of Russia" Diversity 14, no. 10: 859. https://doi.org/10.3390/d14100859
APA StyleChernukha, I., Abdelmanova, A., Kotenkova, E., Kharzinova, V., & Zinovieva, N. A. (2022). Assessing Genetic Diversity and Searching for Selection Signatures by Comparison between the Indigenous Livni and Duroc Breeds in Local Livestock of the Central Region of Russia. Diversity, 14(10), 859. https://doi.org/10.3390/d14100859