
Effect of Polytomy on the Parameter Estimation and Goodness of Fit of Phylogenetic Linear Regression Models for Trait Evolution



Abstract
:1. Introduction
1.1. Linear Regression Analysis
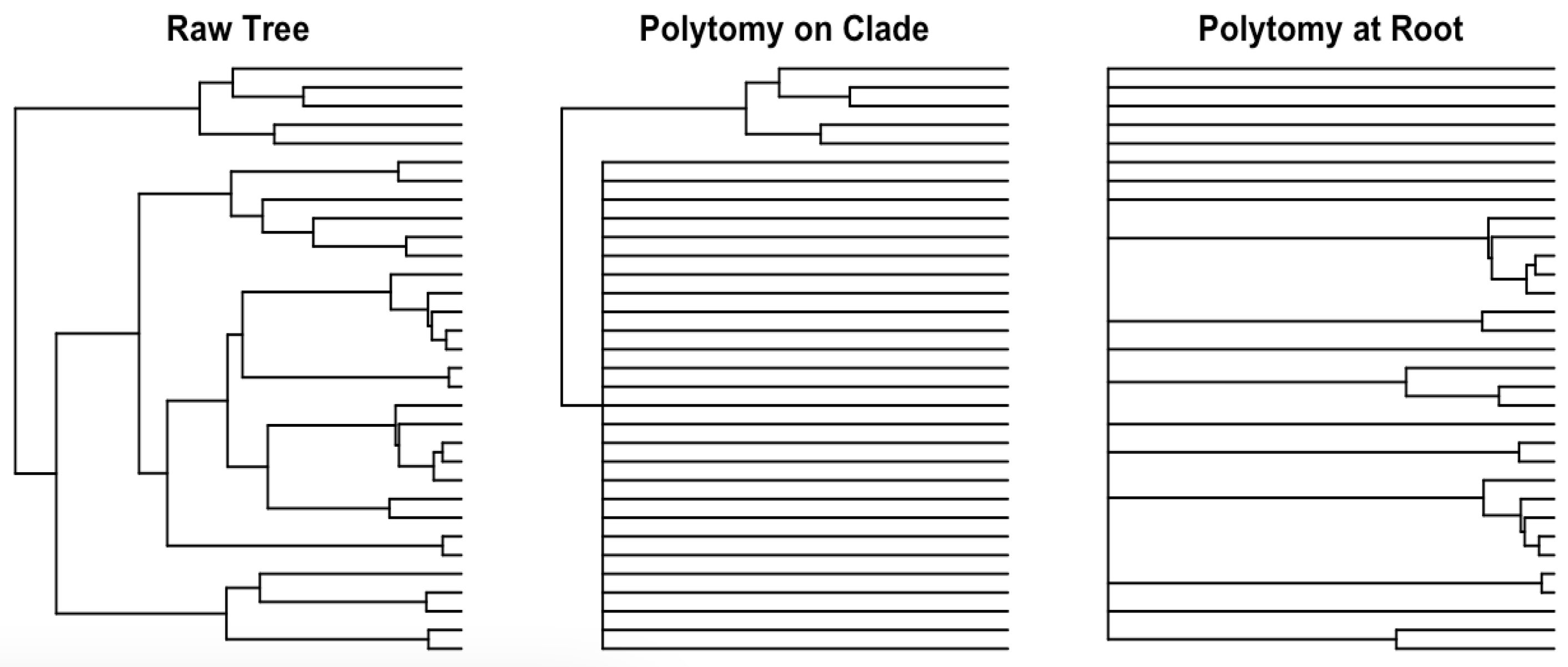
1.2. Tree Polytomy
2. Methods
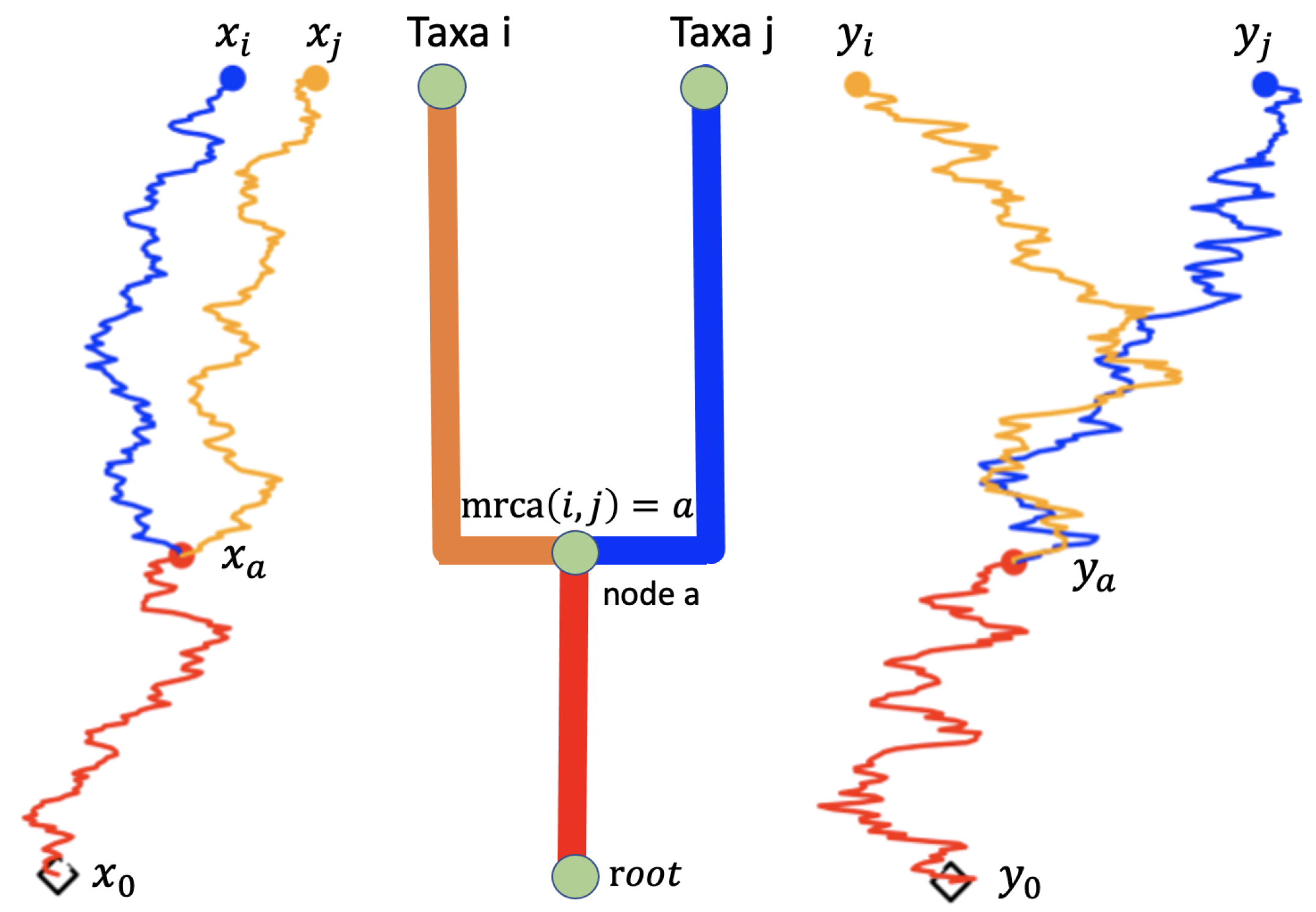
2.1. Models Using Continuous Random Process on Trees for Trait Evolution
2.2. Assessment through Extensive Simulation
3. Results
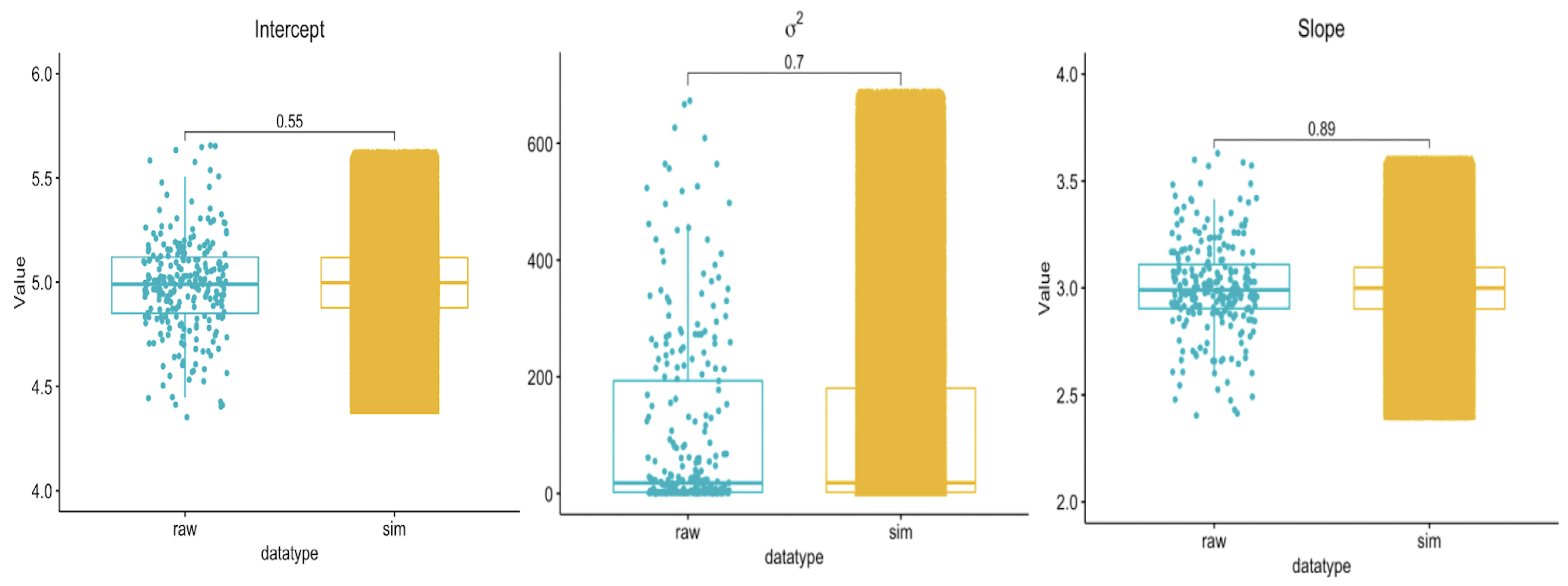
3.1. Overall Estimate
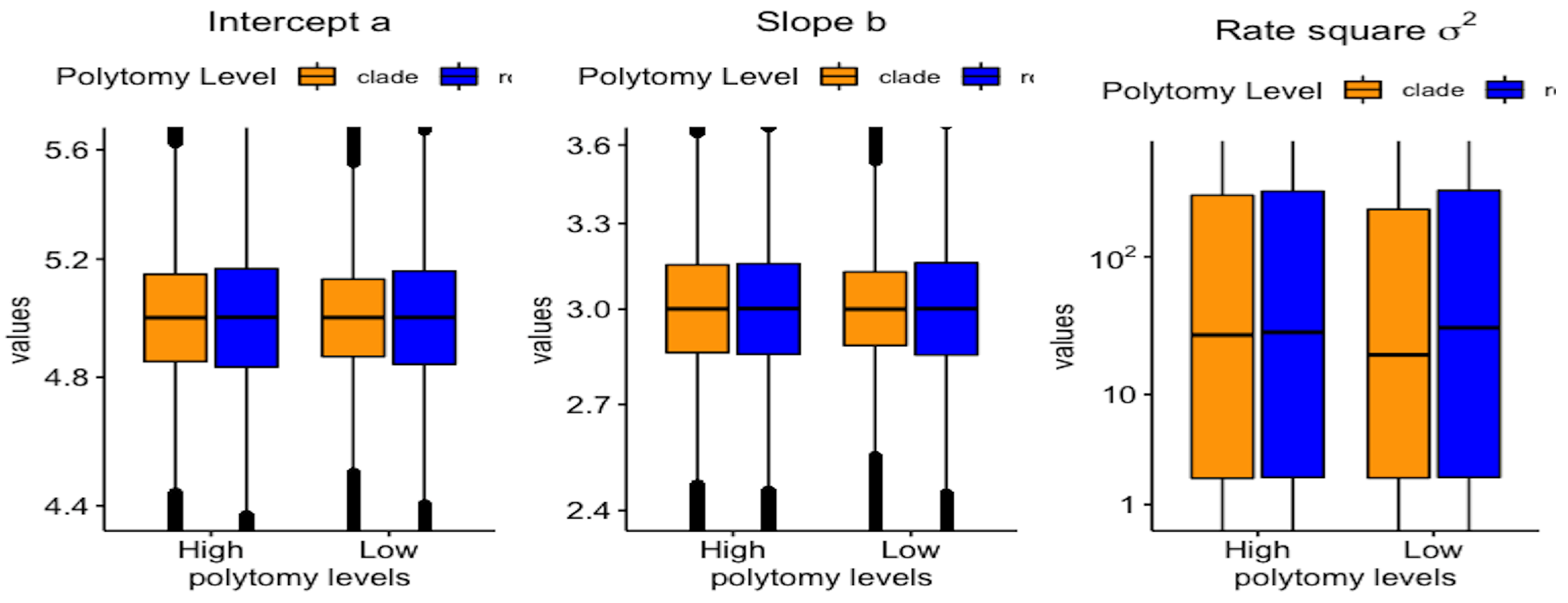
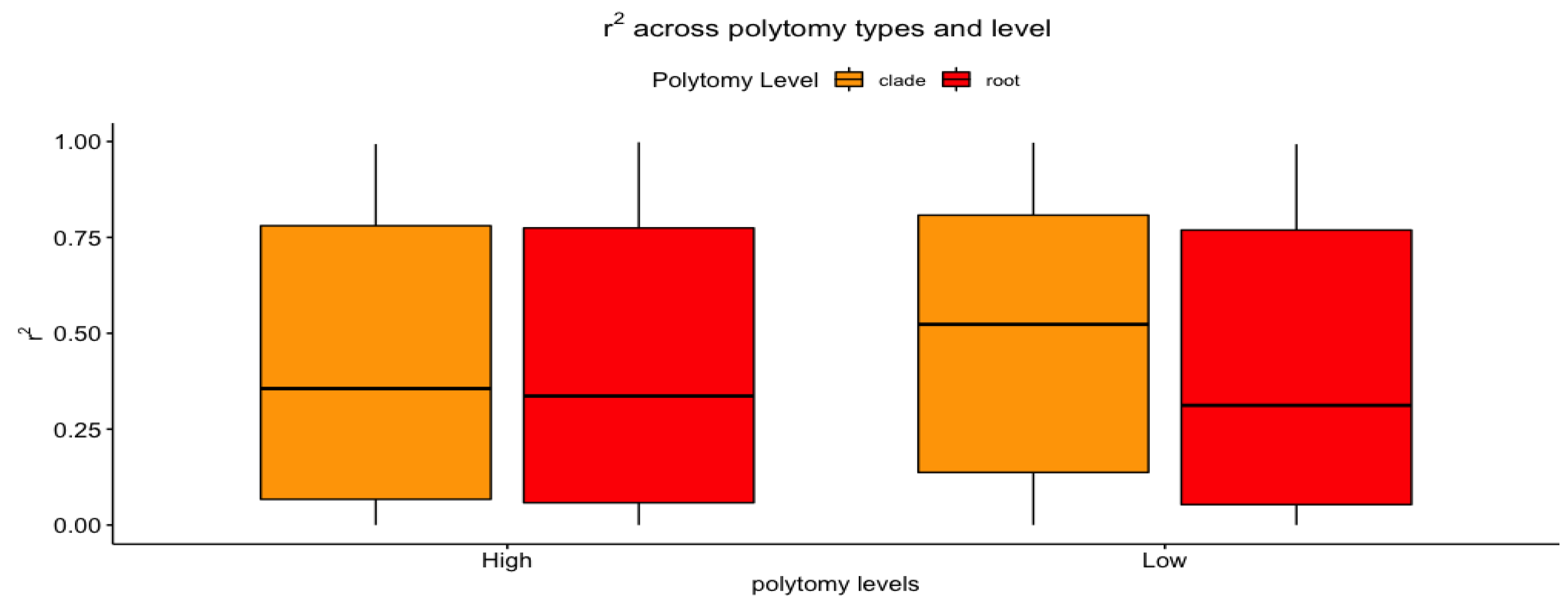
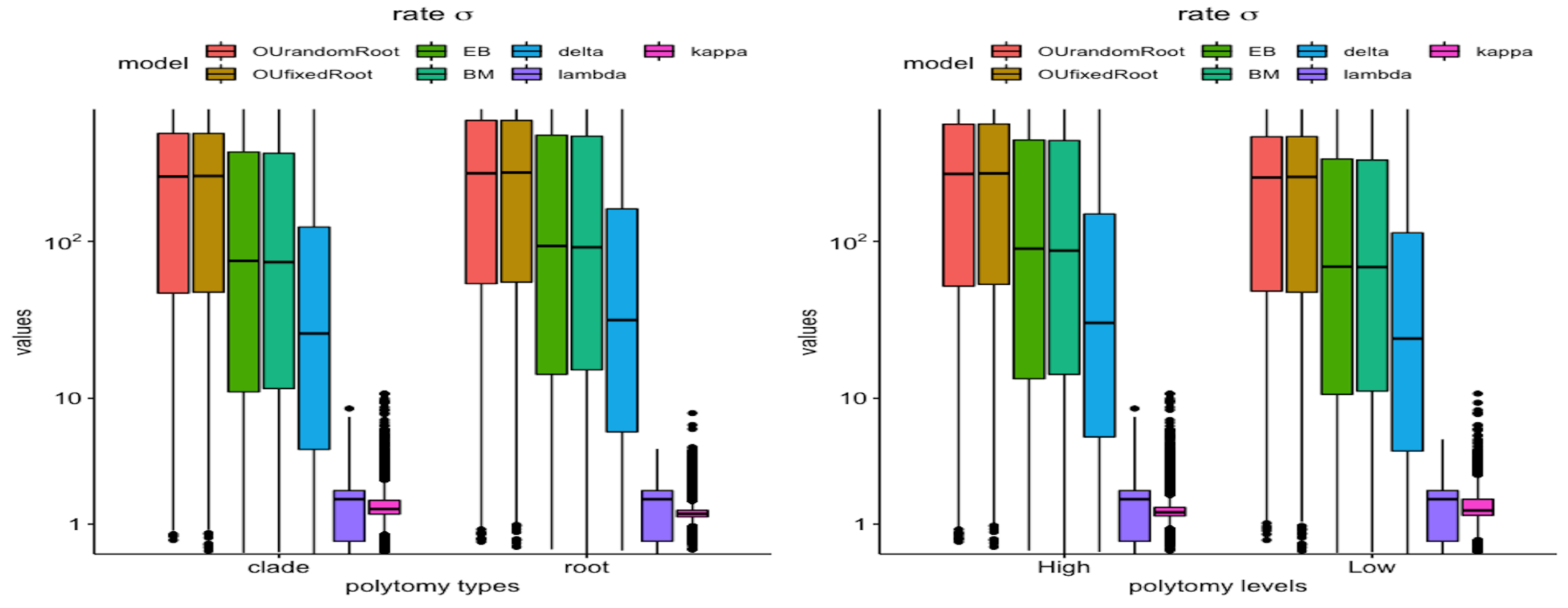
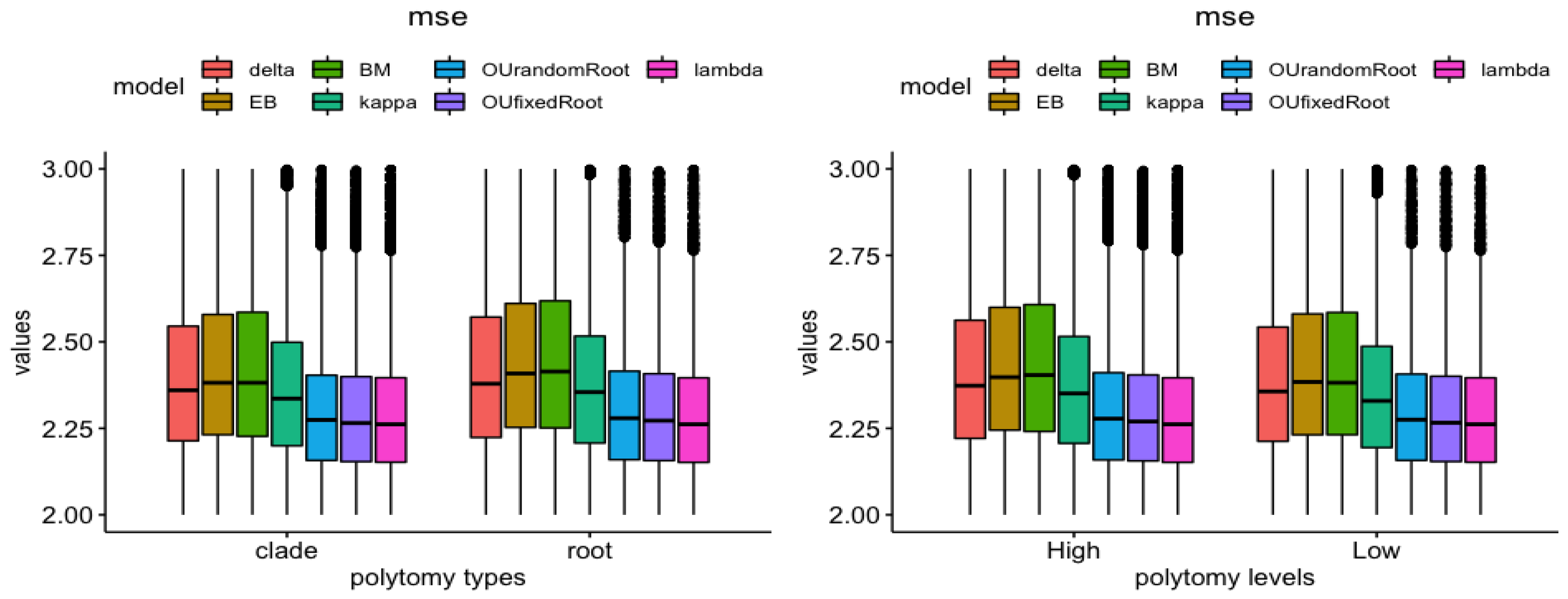
3.2. Polytomy Type vs. Polytomy Level
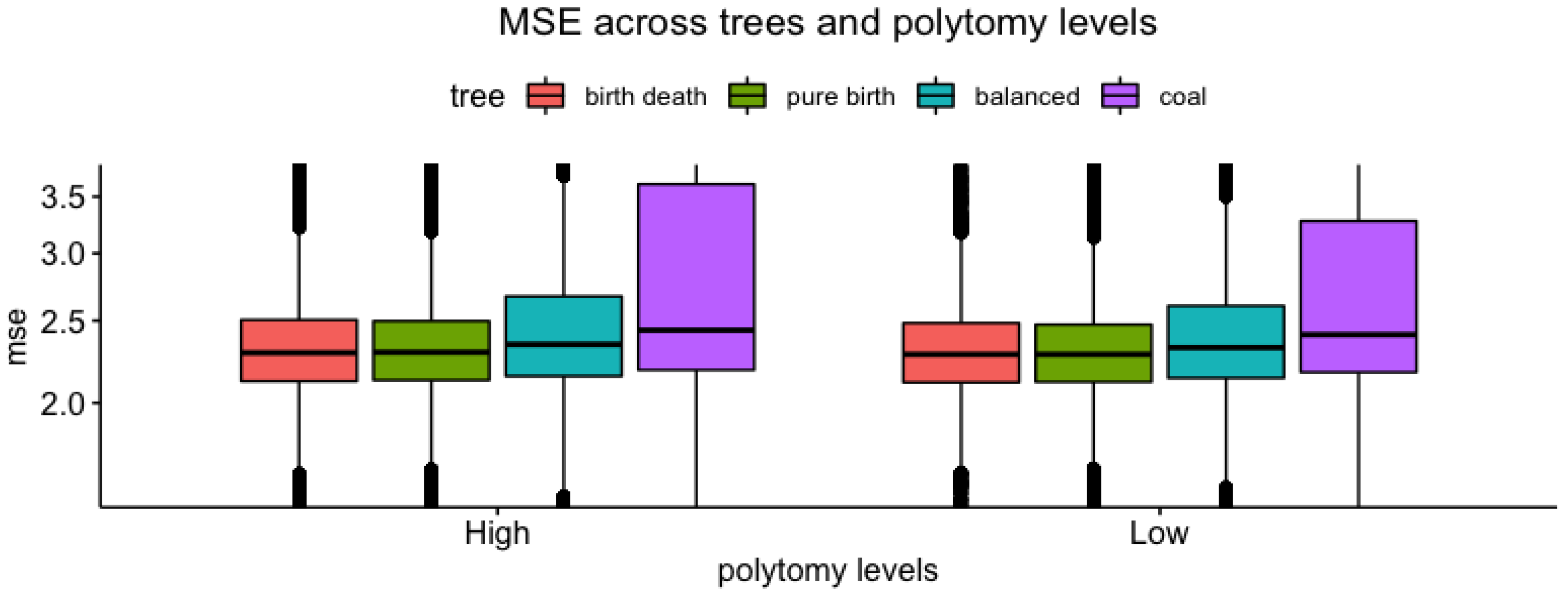
3.3. Tree vs. Measures
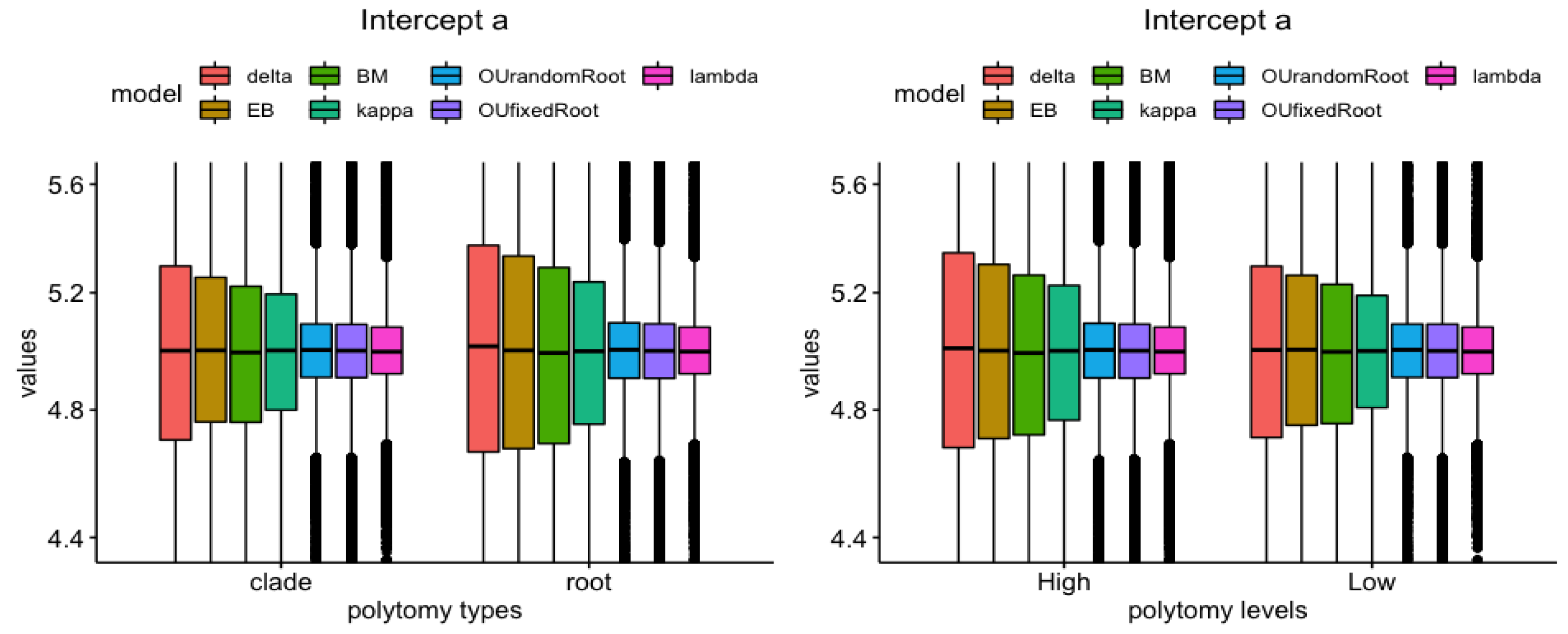
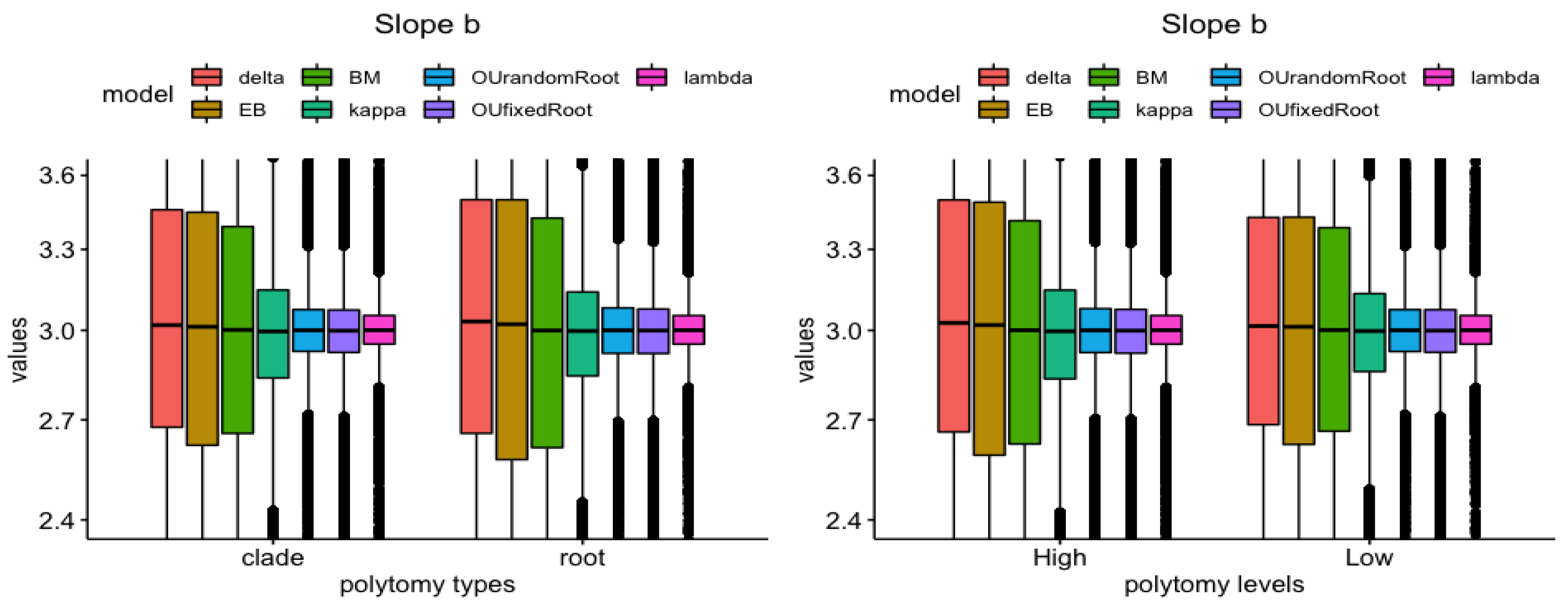
3.4. Model vs. Measure
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Script and Link for Reproducing Result
- 1
- Figure 1: https://tonyjhwueng.info/pcmreg/ttev2.pptx (accessed date: 19 September 2022).
- 2
- Figure 2: https://tonyjhwueng.info/pcmreg/scatterclusterv2.html (accessed date: 19 September 2022).
- 3
- Figure 3: https://tonyjhwueng.info/pcmreg/ttevprog.pptx (accessed date: 19 September 2022).
- 4
- Figure 4: https://tonyjhwueng.info/pcmreg/bm1bm2reg.html (accessed date: 19 September 2022).
- 5
- Figure 5: https://tonyjhwueng.info/pcmreg/bmpathv3_outpathv3_ebpathv2.html (accessed date: 19 September 2022).
- 6
- Figure 6: https://tonyjhwueng.info/pcmreg/makepolytree.html (accessed date: 19 September 2022).
- 7
- Figure 7: https://tonyjhwueng.info/pcmreg/mainsimSummarWrapabsig2.html (accessed date: 19 September 2022).
- 8
- Table 1 and Table 2: https://tonyjhwueng.info/pcmreg/mainsimSummarWrapTable.html (accessed date: 19 September 2022).
- 9
- Figure 8, Figure 9, Figure 10, Figure 11, Figure 12, Figure 13, Figure 14, Figure 15, Figure 16, Figure 17, Figure 18, Figure 19 and Figure 20:https://tonyjhwueng.info/pcmreg/mainsimSummarWrapBoxplotv4.html (accessed date: 19 September 2022).
Appendix A.2. Database for Accessing Comparative Data and Tree

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Logo | Database | Reference | Link |
|---|---|---|---|
 | AmphibiaWeb | [22] | https://amphibiaweb.org/ (accessed date: 19 September 2022). |
 | The Reptile Database | [23] | http://www.reptile-database.org/ (accessed date: 19 September 2022). |
 | GLAD | [24] | http://globalants.org/ (accessed date: 19 September 2022). |
 | DateLife | [71] | http://datelife.opentreeoflife.org (accessed date: 19 September 2022). |
 | EzBioCloud | [72] | https://www.ezbiocloud.net (accessed date: 19 September 2022). |
 | FishBase | [25] | https://www.fishbase.se/ (accessed date: 19 September 2022). |
 | Open Tree of Life | [32] | https://tree.opentreeoflife.org/ (accessed date: 19 September 2022). |
 | PhylomeDB | [73] | http://phylomedb.org/ (accessed date: 19 September 2022). |
 | PHYLOtastic | [28] | https://phylotastic.org/ (accessed date: 19 September 2022). |
 | Traitbase | [74] | https://traitbase.info/ (accessed date: 19 September 2022). |
 | TreeBASE | [75] | https://www.treebase.org/ (accessed date: 19 September 2022). |
 | Treefam | [26] | http://www.treefam.org (accessed date: 19 September 2022). |
 | Tree of Life Web Project | [29] | http://tolweb.org/tree/ (accessed date: 19 September 2022). |
 | The Open Traits Network | [30] | https://opentraits.org/ (accessed date: 19 September 2022). |
 | TRY Plant Trait Database | [27] | https://www.try-db.org/ (accessed date: 19 September 2022). |
Appendix A.3. Covariance Matrix
References
- Garamszegi, L.Z. Modern Phylogenetic Comparative Methods and Their Application in Evolutionary Biology: Concepts and Practice; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Paradis, E. An introduction to the phylogenetic comparative method. In Modern Phylogenetic Comparative Methods and Their Application in Evolutionary Biology; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3–18. [Google Scholar]
- Martins, E.P.; Martins, E.P. Phylogenies and the Comparative Method in Animal Behavior; Oxford University Press on Demand: Oxford, UK, 1996. [Google Scholar]
- Choudhuri, S. Bioinformatics for Beginners: Genes, Genomes, Molecular Evolution, Databases and Analytical Tools; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [Green Version]
- Furness, A.I.; Capellini, I. The evolution of parental care diversity in amphibians. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Woolley, C.H.; Thompson, J.R.; Wu, Y.H.; Bottjer, D.J.; Smith, N.D. A biased fossil record can preserve reliable phylogenetic signal. Paleobiology 2022, 48, 1–16. [Google Scholar] [CrossRef]
- Polly, P.D.; Stayton, C.T.; Dumont, E.R.; Pierce, S.E.; Rayfield, E.J.; Angielczyk, K.D. Combining geometric morphometrics and finite element analysis with evolutionary modeling: Towards a synthesis. J. Vertebr. Paleontol. 2016, 36, e1111225. [Google Scholar] [CrossRef] [Green Version]
- Davies, E.L.; Arbuckle, K. Coevolution of snake venom toxic activities and diet: Evidence that ecological generalism favours toxicological diversity. Toxins 2019, 11, 711. [Google Scholar] [CrossRef] [Green Version]
- Krüger, O. The evolution of reversed sexual size dimorphism in hawks, falcons and owls: A comparative study. Evol. Ecol. 2005, 19, 467–486. [Google Scholar] [CrossRef]
- Pagel, M.; Meade, A.; Barker, D. Bayesian estimation of ancestral character states on phylogenies. Syst. Biol. 2004, 53, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.; Jhwueng, D.C.; Boettiger, C.; O’Meara, B. Modeling stabilizing selection: Expanding the Ornstein-Uhlenbeck model of adaptive evolution. Evolution 2012, 66, 2369–2383. [Google Scholar] [CrossRef]
- Besnard, F.; Picao-Osorio, J.; Dubois, C.; Félix, M.A. A broad mutational target explains a fast rate of phenotypic evolution. Elife 2020, 9, e54928. [Google Scholar] [CrossRef]
- Pannetier, T.; Martinez, C.; Bunnefeld, L.; Etienne, R.S. Branching patterns in phylogenies cannot distinguish diversity-dependent diversification from time-dependent diversification. Evolution 2021, 75, 25–38. [Google Scholar] [CrossRef]
- Xue, B.; Guo, X.; Landis, J.; Sun, M.; Tang, C.; Soltis, P.; Soltis, D.; Saunders, R. Accelerated diversification correlated with functional traits shapes extant diversity of the early divergent angiosperm family Annonaceae. Mol. Phylogenet. Evol. 2020, 142, 106659. [Google Scholar] [CrossRef]
- O’Meara, B.C. Evolutionary inferences from phylogenies: A review of methods. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 267–285. [Google Scholar] [CrossRef]
- Vasconcelos, T.; O’Meara, B.C.; Beaulieu, J.M. A flexible method for estimating tip diversification rates across a range of speciation and extinction scenarios. Evolution 2022, 76, 1420–1433. [Google Scholar] [CrossRef]
- Duchen, P.; Alfaro, M.L.; Rolland, J.; Salamin, N.; Silvestro, D. On the effect of asymmetrical trait inheritance on models of trait evolution. Syst. Biol. 2021, 70, 376–388. [Google Scholar] [CrossRef]
- Smaers, J.B.; Dechmann, D.K.; Goswami, A.; Soligo, C.; Safi, K. Comparative analyses of evolutionary rates reveal different pathways to encephalization in bats, carnivorans, and primates. Proc. Natl. Acad. Sci. USA 2012, 109, 18006–18011. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, A.P.; Santos, B.A.; Betancur-R, R. Does Genome Size Increase with Water Depth in Marine Fishes? Evolution 2022, 76, 1578–1589. [Google Scholar] [CrossRef]
- Bickford, D.; Lohman, D.; Navjot, S.; Ng, P.; Meier, R.; Winker, K.; Ingram, K.; Feinberg, J.; Newman, C.; Watkins-Colwell, G.; et al. AmphibiaWeb. 2017. Available online: http://amphibiaweb.org (accessed on 19 September 2022).
- Uetz, P.; Koo, M.S.; Aguilar, R.; Brings, E.; Catenazzi, A.; Chang, A.; Wake, D. A quarter century of reptile and amphibian databases. Herpetol. Rev. 2021, 52, 246–255. [Google Scholar]
- Parr, C.L.; Dunn, R.R.; Sanders, N.J.; Weiser, M.D.; Photakis, M.; Bishop, T.R.; Fitzpatrick, M.C.; Arnan, X.; Baccaro, F.; Brandão, C.R.; et al. GlobalAnts: A new database on the geography of ant traits (Hymenoptera: Formicidae). Insect Conserv. Divers. 2017, 10, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Pauly, D.; Froese, R. FishBase. 2010. Available online: https://www.fishbase.se/search.php (accessed on 19 September 2022).
- Schreiber, F.; Patricio, M.; Muffato, M.; Pignatelli, M.; Bateman, A. TreeFam v9: A new website, more species and orthology-on-the-fly. Nucleic Acids Res. 2014, 42, D922–D925. [Google Scholar] [CrossRef] [Green Version]
- Kattge, J.; Bönisch, G.; Díaz, S.; Lavorel, S.; Prentice, I.C.; Leadley, P.; Tautenhahn, S.; Werner, G.D.; Aakala, T.; Abedi, M.; et al. TRY plant trait database–enhanced coverage and open access. Glob. Chang. Biol. 2020, 26, 119–188. [Google Scholar] [CrossRef] [PubMed]
- Stoltzfus, A.; Lapp, H.; Matasci, N.; Deus, H.; Sidlauskas, B.; Zmasek, C.M.; Vaidya, G.; Pontelli, E.; Cranston, K.; Vos, R.; et al. Phylotastic! Making tree-of-life knowledge accessible, reusable and convenient. BMC Bioinform. 2013, 14, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Maddison, D.R.; Schulz, K.S.; Maddison, W.P. The tree of life web project. Zootaxa 2007, 1668, 19–40. [Google Scholar] [CrossRef]
- Gallagher, R.; Falster, D.; Maitner, B.; Salguero-Gomez, R.; Vandvik, V.; Pearse, W.; Enquist, B. Open Science principles for accelerating trait-based science across the Tree of Life. Nat. Ecol. Evol. 2020, 4, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Kleyer, M.; Bekker, R.; Knevel, I.; Bakker, J.; Thompson, K.; Sonnenschein, M.; Poschlod, P.; Van Groenendael, J.; Klimeš, L.; Klimešová, J.; et al. The LEDA Traitbase: A database of life-history traits of the Northwest European flora. J. Ecol. 2008, 96, 1266–1274. [Google Scholar] [CrossRef]
- Michonneau, F.; Brown, J.W.; Winter, D.J. rotl: An R package to interact with the Open Tree of Life data. Methods Ecol. Evol. 2016, 7, 1476–1481. [Google Scholar] [CrossRef]
- McTavish, E.J.; Sánchez-Reyes, L.L.; Holder, M.T. OpenTree: A Python package for Accessing and Analyzing data from the Open Tree of Life. Syst. Biol. 2021, 70, 1295–1301. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Bueno, A.; Dopazo, J.; Gabaldón, T. PhylomeDB: A database for genome-wide collections of gene phylogenies. Nucleic Acids Res. 2007, 36, D491–D496. [Google Scholar] [CrossRef]
- Smith, S.D.; Ané, C.; Baum, D.A. The role of pollinator shifts in the floral diversification of Iochroma (Solanaceae). Evol. Int. J. Org. Evol. 2008, 62, 793–806. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- O’Meara, B. CRAN Task View: Phylogenetics, Especially Comparative Methods. 2020. Available online: https://cran.r-project.org/web/views/Phylogenetics.html (accessed on 2 November 2022).
- Ané, C. Analysis of comparative data with hierarchical autocorrelation. Ann. Appl. Stat. 2008, 2, 1078–1102. [Google Scholar] [CrossRef]
- Revell, L.J. Phylogenetic signal and linear regression on species data. Methods Ecol. Evol. 2010, 1, 319–329. [Google Scholar] [CrossRef]
- Stone, E.A. Why the phylogenetic regression appears robust to tree misspecification. Syst. Biol. 2011, 60, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.; Burnham, K. Model Selection and Multi-Model Inference, 2nd ed.; Springer: New York, NY, USA, 2004; Volume 63, p. 10. [Google Scholar]
- Felsenstein, J. Phylogeny and the comparative method. Am. Nat. 1985, 125, 1–15. [Google Scholar] [CrossRef]
- Hansen, T.F. Stabilizing selection and the comparative analysis of adaptation. Evolution 1997, 51, 1341–1351. [Google Scholar] [CrossRef]
- Harmon, L.J.; Losos, J.B.; Jonathan Davies, T.; Gillespie, R.G.; Gittleman, J.L.; Bryan Jennings, W.; Kozak, K.H.; McPeek, M.A.; Moreno-Roark, F.; Near, T.J.; et al. Early bursts of body size and shape evolution are rare in comparative data. Evol. Int. J. Org. Evol. 2010, 64, 2385–2396. [Google Scholar] [CrossRef]
- Pagel, M. Inferring the historical patterns of biological evolution. Nature 1999, 401, 877. [Google Scholar] [CrossRef]
- Adams, D.C.; Collyer, M.L. Multivariate phylogenetic comparative methods: Evaluations, comparisons, and recommendations. Syst. Biol. 2018, 67, 14–31. [Google Scholar] [CrossRef]
- Billingsley, P. Probability and Measure; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Hansen, T.; Martins, E. Translating between microevolutionary process and macroevolutionary patterns: The correlation structure of interspecific data. Evolution 1996, 50, 1404–1417. [Google Scholar] [CrossRef]
- Butler, M.; King, A. Phylogenetic comparative analysis: A modeling approach for adaptive evolution. Am. Nat. 2004, 164, 683–695. [Google Scholar] [CrossRef]
- Housworth, E.A.; Martins, E.P.; Lynch, M. The phylogenetic mixed model. Am. Nat. 2004, 163, 84–96. [Google Scholar] [CrossRef] [Green Version]
- Harmon, L.J. Phylogenetic Comparative Methods. 2019. Available online: https://lukejharmon.github.io/pcm/ (accessed on 2 November 2022).
- Revell, L.J.; Harmon, L.J. Phylogenetic Comparative Methods in R; Princeton University Press: Princeton, NJ, USA, 2022. [Google Scholar]
- Hansen, T.; Pienaar, J.; Orzack, S. A Comparative Method for Studying Adaptation to a Randomly Evolving Environment. Evolution 2008, 62, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.S.T.; Ane, C.; Lachlan, R.; Tarpinian, K.; Feldman, R.; Yu, Q.; van der Bijl, W.; Maspons, J.; Vos, R.; Ho, M.L.S.T. Package ‘Phylolm’. 2016. Available online: http://cran. r-project.org/web/packages/phylolm/index.html (accessed on 2 February 2021).
- Castiglione, S.; Serio, C.; Mondanaro, A.; Melchionna, M.; Carotenuto, F.; Di Febbraro, M.; Profico, A.; Tamagnini, D.; Raia, P. Ancestral state estimation with phylogenetic ridge regression. Evol. Biol. 2020, 47, 220–232. [Google Scholar] [CrossRef]
- Smith, M.R. TreeTools: Create, Modify and Analyse Phylogenetic Trees. Comprehensive R Archive Network. 2019. R Package Version 1.7.3. Available online: https://doi.org/10.5281/zenodo.3522725 (accessed on 19 September 2022).
- Bossio, M.C.; Cuervo, E.C. Gamma regression models with the Gammareg R package. Comun. En Estadística 2015, 8, 211–223. [Google Scholar] [CrossRef]
- Ives, A.R.; Garland, T., Jr. Phylogenetic logistic regression for binary dependent variables. Syst. Biol. 2010, 59, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribari-Neto, F.; Zeileis, A. Beta regression in R. J. Stat. Softw. 2010, 34, 1–24. [Google Scholar] [CrossRef] [Green Version]
- O’Meara, B.; Ané, C.; Sanderson, M.; Wainwright, P. Testing different rates of continuous trait evolution using likelihood. Evolution 2006, 60, 922–933. [Google Scholar] [CrossRef]
- Manceau, M.; Lambert, A.; Morlon, H. A unifying comparative phylogenetic framework including traits coevolving across interacting lineages. Syst. Biol. 2017, 66, 551–568. [Google Scholar] [CrossRef]
- Jhwueng, D.C.; Maroulas, V. Phylogenetic ornstein–uhlenbeck regression curves. Stat. Probab. Lett. 2014, 89, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Jhwueng, D.C.; Maroulas, V. Adaptive trait evolution in random environment. J. Appl. Stat. 2016, 43, 2310–2324. [Google Scholar] [CrossRef] [Green Version]
- Bastide, P.; Solís-Lemus, C.; Kriebel, R.; William Sparks, K.; Ané, C. Phylogenetic comparative methods on phylogenetic networks with reticulations. Syst. Biol. 2018, 67, 800–820. [Google Scholar] [CrossRef] [PubMed]
- Jhwueng, D.C. Reduced drought tolerance during domestication and the evolution of weediness results from tolerance–growth trade-offs. Evol. Int. J. Org. Evol. 2012, 66, 3803–3814. [Google Scholar]
- Uyeda, J.C.; Harmon, L.J. A novel Bayesian method for inferring and interpreting the dynamics of adaptive landscapes from phylogenetic comparative data. Syst. Biol. 2014, 63, 902–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villemereuil, P.d.; Wells, J.A.; Edwards, R.D.; Blomberg, S.P. Bayesian models for comparative analysis integrating phylogenetic uncertainty. BMC Evol. Biol. 2012, 12, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anscombe, F.J. Graphs in statistical analysis. Am. Stat. 1973, 27, 17–21. [Google Scholar]
- Revell, L.J.; Schliep, K.; Valderrama, E.; Richardson, J.E. Graphs in phylogenetic comparative analysis: Anscombe’s quartet revisited. Methods Ecol. Evol. 2018, 9, 2145–2154. [Google Scholar] [CrossRef]
- Liang, X.; Wang, Z.; Zhou, Z.; Huang, Z.; Zhou, J.; Cen, K. Up-to-date life cycle assessment and comparison study of clean coal power generation technologies in China. J. Clean. Prod. 2013, 39, 24–31. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Capella-Gutierrez, S.; Pryszcz, L.P.; Marcet-Houben, M.; Gabaldon, T. PhylomeDB v4: Zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 2014, 42, D897–D902. [Google Scholar] [CrossRef] [Green Version]
- Adler, P.B.; Fajardo, A.; Kleinhesselink, A.R.; Kraft, N.J. Trait-based tests of coexistence mechanisms. Ecol. Lett. 2013, 16, 1294–1306. [Google Scholar] [CrossRef]
- Piel, W.H.; Donoghue, M.; Sanderson, M.; Netherlands, L. TreeBASE: A database of phylogenetic information. In Proceedings of the 2nd International Workshop of Species, Copenhagen, Denmark, 22–26 May 2000; Volume 2000. [Google Scholar]




















| Intercept | Slope | AIC | R | MSE | ||
|---|---|---|---|---|---|---|
| Raw | ||||||
| Sim |
| Polytomy Type (Root vs. Clade) | Polytomy Level (High vs. Low) | |
|---|---|---|
| Intercept a | , | , |
| Slope b | , | , |
| , | , | |
| AIC | , | , |
| , * | , | |
| MSE | , | , |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jhwueng, D.-C.; Liu, F.-C. Effect of Polytomy on the Parameter Estimation and Goodness of Fit of Phylogenetic Linear Regression Models for Trait Evolution. Diversity 2022, 14, 942. https://doi.org/10.3390/d14110942
Jhwueng D-C, Liu F-C. Effect of Polytomy on the Parameter Estimation and Goodness of Fit of Phylogenetic Linear Regression Models for Trait Evolution. Diversity. 2022; 14(11):942. https://doi.org/10.3390/d14110942
Chicago/Turabian StyleJhwueng, Dwueng-Chwuan, and Feng-Chi Liu. 2022. "Effect of Polytomy on the Parameter Estimation and Goodness of Fit of Phylogenetic Linear Regression Models for Trait Evolution" Diversity 14, no. 11: 942. https://doi.org/10.3390/d14110942
APA StyleJhwueng, D. -C., & Liu, F. -C. (2022). Effect of Polytomy on the Parameter Estimation and Goodness of Fit of Phylogenetic Linear Regression Models for Trait Evolution. Diversity, 14(11), 942. https://doi.org/10.3390/d14110942