
Characterization of the Complete Chloroplast Genome of the Dragonhead Herb, Dracocephalumheterophyllum (Lamiaceae), and Comparative Analyses with Related Species


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. Chloroplast Genome Sequencing and Assembly
2.3. Annotation and Analysis of Chloroplast Genomes
2.4. Single Sequence Repeats (SSR) and Relative Synonymous Codon Usage (RSCU) Analysis
2.5. Complete Chloroplast Genome of Comparison Analysis
2.6. Analysis of Synonymous (Ks) and Non-Synonymous (Ka) Substitution Rate
2.7. Phylogenetic Analysis
3. Results
3.1. General Characterization of Chloroplast Genome
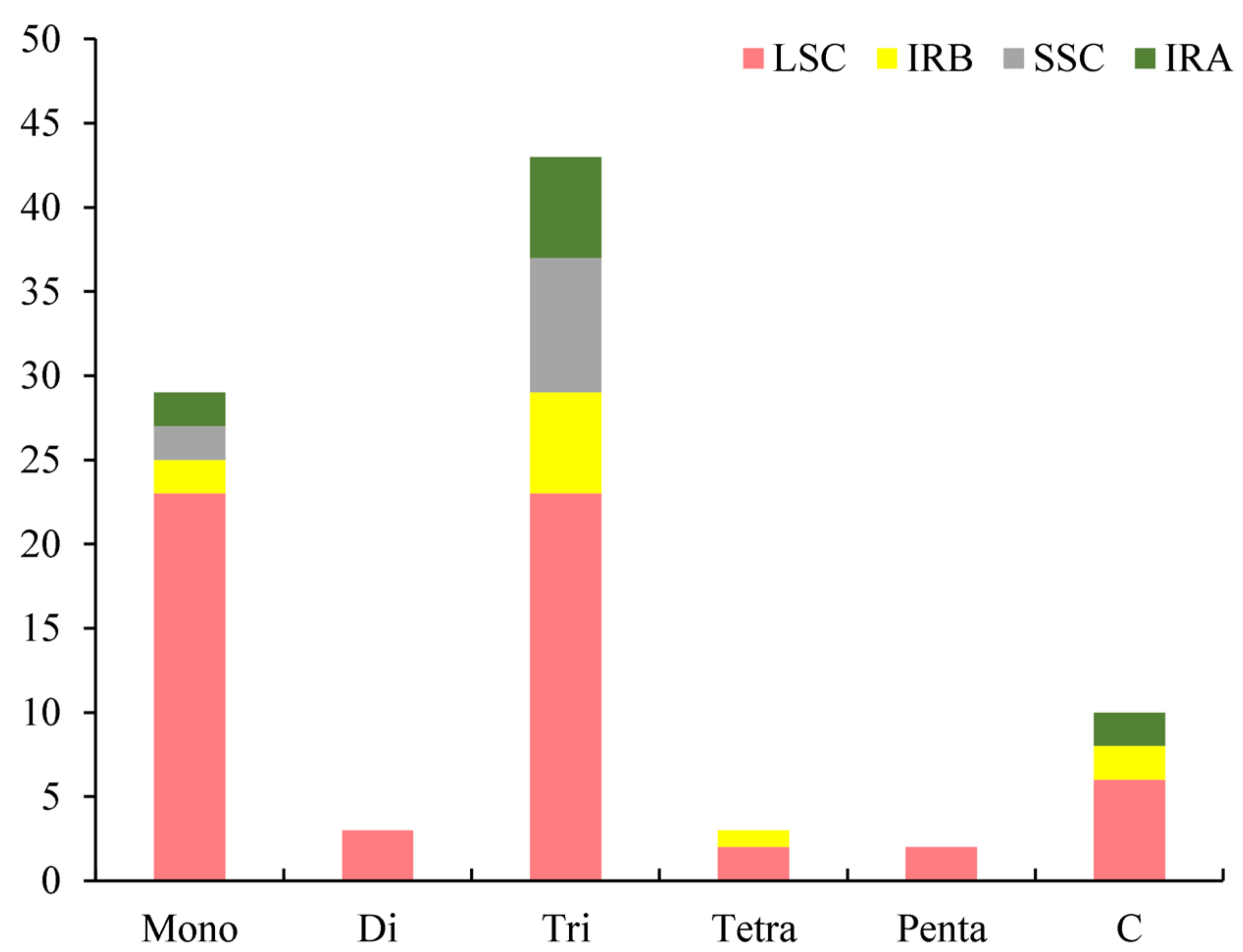
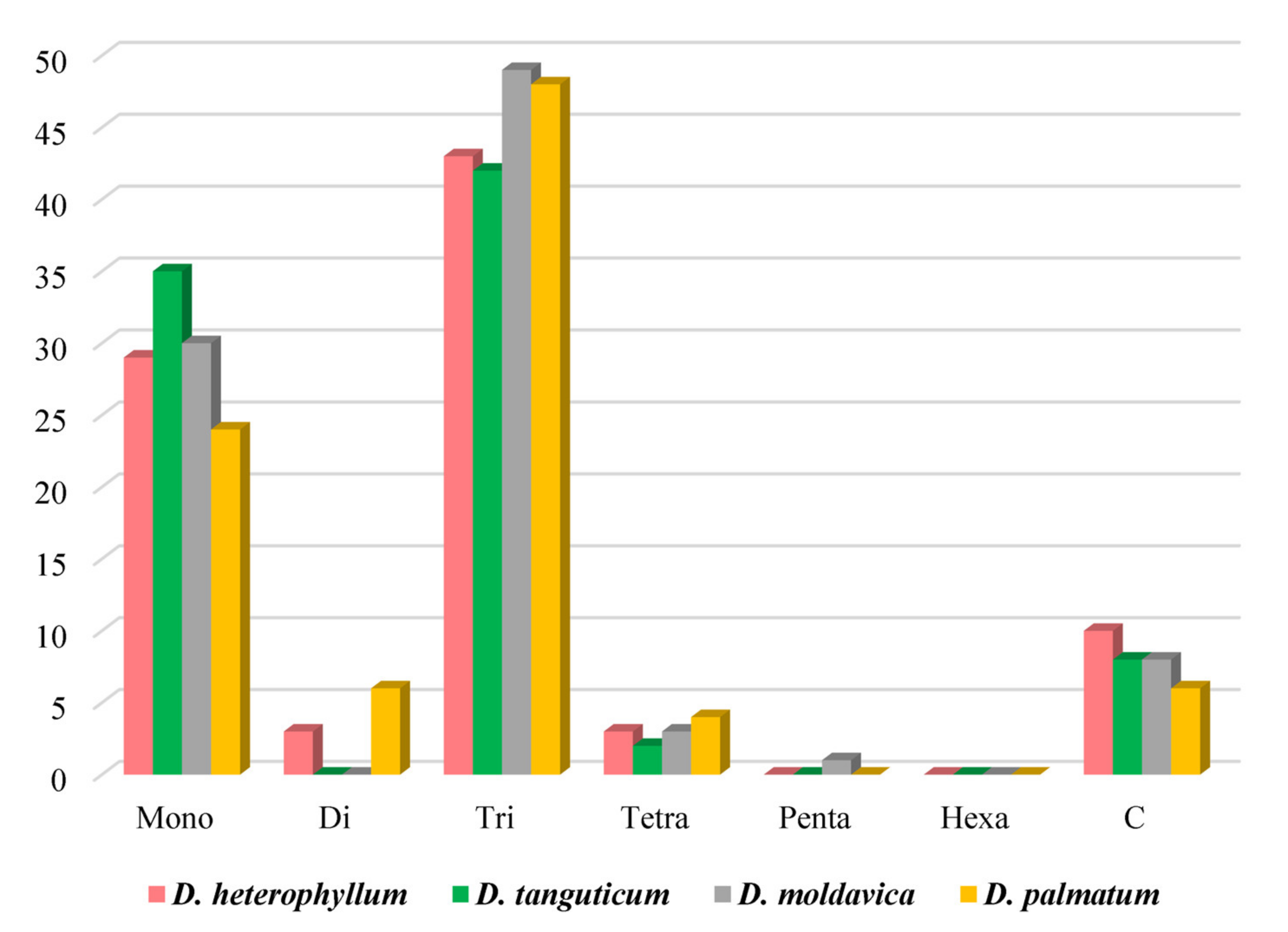
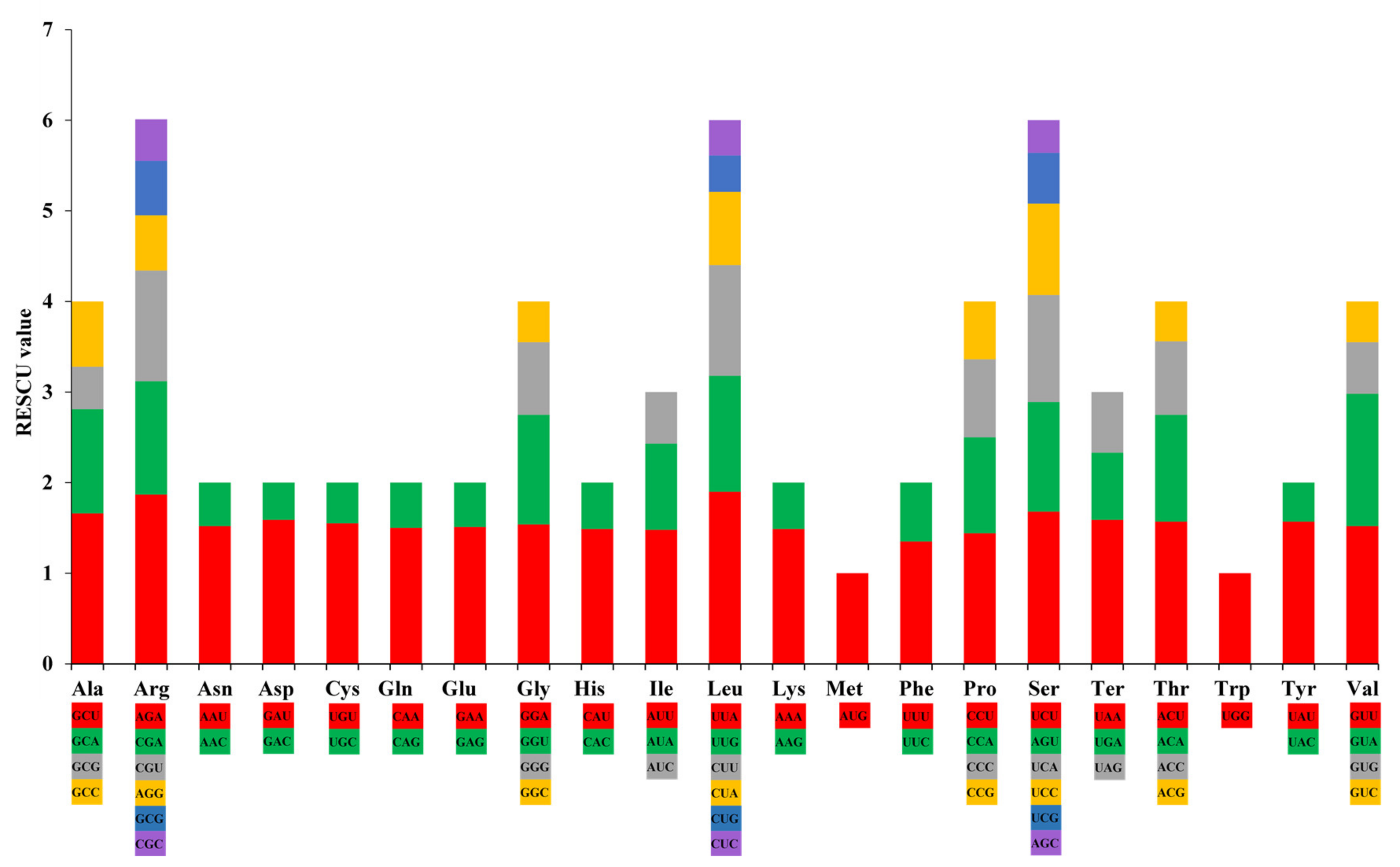
3.2. Analysis of Simple Sequence Repeat (SSR) and Codon Usage
3.3. IR Contraction and Expansion
3.4. Comparisons of the cp Genome between D. heterophyllum and other Lamiaceae species
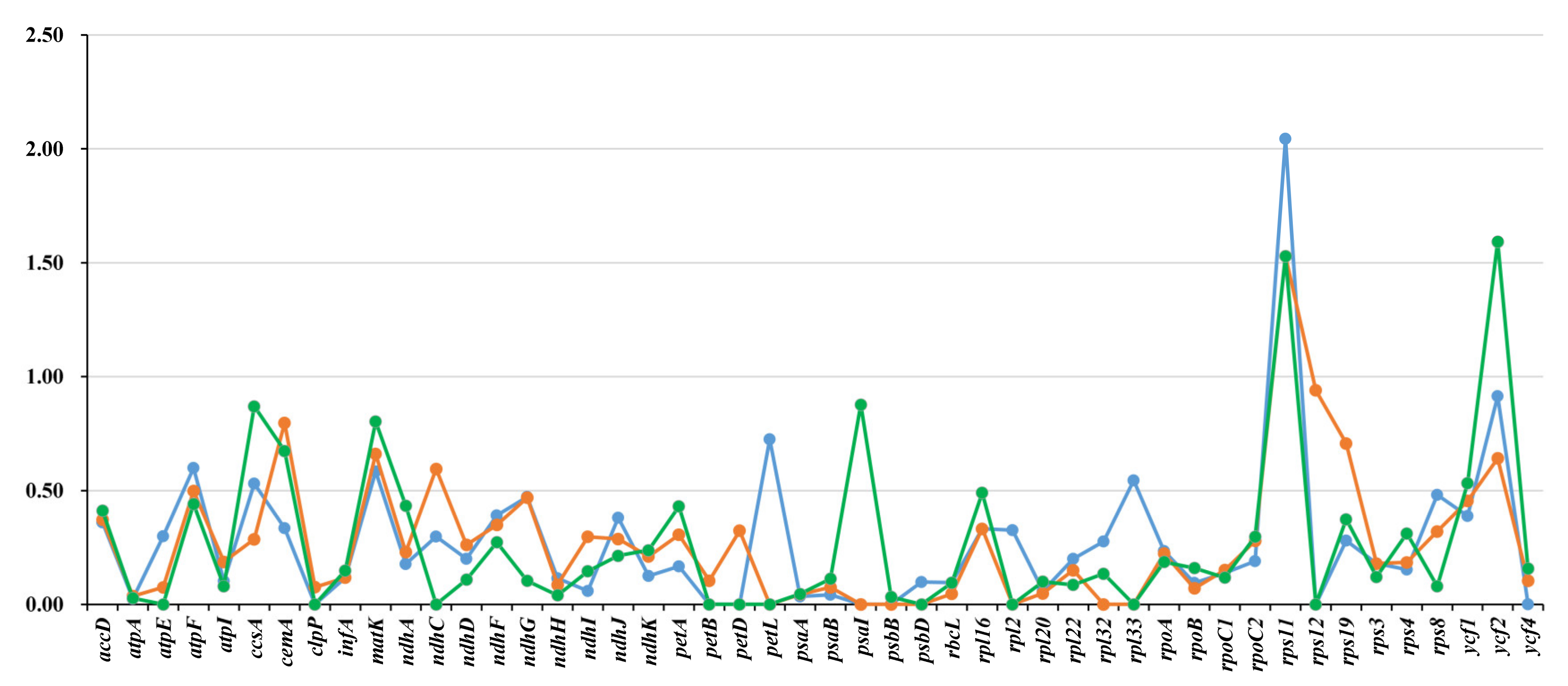
3.5. Characterization of Substitution Rates
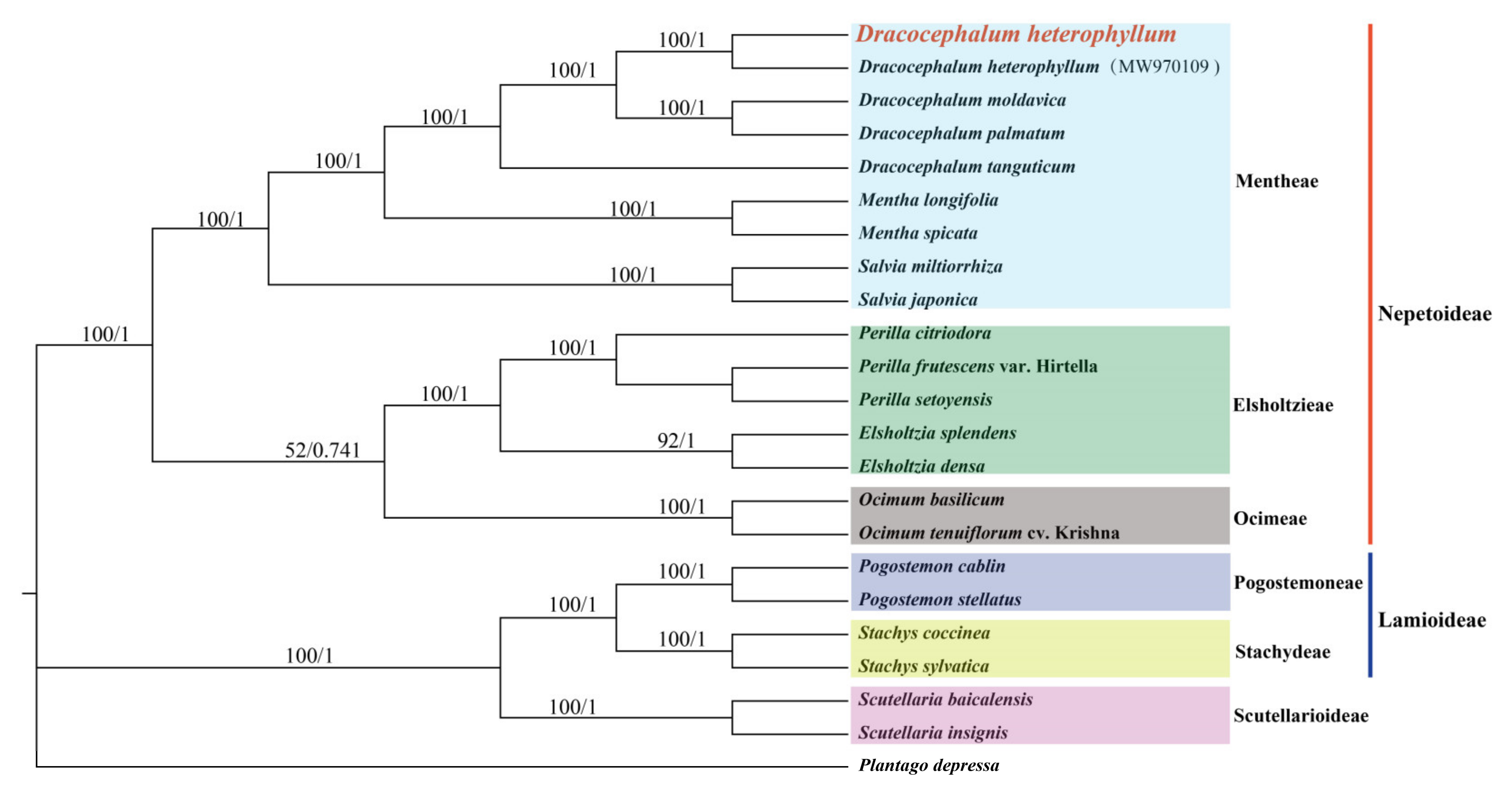
3.6. Evolutionary and Phylogenetic Analysis
4. Discussion
4.1. Organization and Features of cp Genomes
4.2. Simple Sequence Repeats (SSRs) and Codon Usage Analysis
4.3. IR Expansion/Contraction of cp Genomes
4.4. Comparative Genomes and Characterization of Substitution Rates
4.5. Phylogenetic Analysis Based on cp Genomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Li, B.; Cantino, P.D.; Olmstead, R.G.; Bramley, G.L.C.; Xiang, C.L.; Ma, Z.H.; Tan, Y.H.; Zhang, D.X. A large-scale chloroplast phylogeny of the Lamiaceae sheds new light on its subfamilial classification. Sci. Rep. 2016, 6, 34343. [Google Scholar] [CrossRef] [Green Version]
- Bunsawat, J.; Elliott, N.E.; Hertweck, K.L.; Sproles, E.; Alice, L.A. Phylogenetics of Mentha (Lamiaceae): Evidence from chloroplast DNA sequences. Syst. Bot. 2004, 29, 959–964. [Google Scholar] [CrossRef]
- Wang, L.M. Chemical and Bioactive Studies on Dracocephalum heterophyllum of the Genus Dracocephalum. Ph.D. Thesis, Shandong University, Jinan, China, 2011. [Google Scholar]
- Ren, A.M. Chemical constituents of Dracocephalum heterophyllum and antibacterial activity. Zhong Cao Yao 2011, 42, 664–667. [Google Scholar] [CrossRef]
- Numonov, S.R.; Usmanova, S.K.; Aisa, H.A. A triterpenoid and flavonoids from Dracocephalum heterophyllum. Chem. Nat. Compd. 2013, 48, 1109–1110. [Google Scholar] [CrossRef]
- Zhang, C.J.; Li, H.Y.; Yun, T.; Fu, Y.H.; Liu, C.M.; Gong, B.; Neng, B.J. Chemical composition, antimicrobial and antioxidant activities of the essential oil of Tibetan herbal medicine Dracocephalum heterophyllum Benth. Nat. Prod. Res. 2008, 22, 1–11. [Google Scholar] [CrossRef]
- Editorial Board of Forage Flora of China. Forage Flora of China, 1st ed.; China Agriculture Science and Technology: Beijing, China, 1992; pp. 471–473. [Google Scholar]
- Raman, G.; Park, V.; Kwak, M.; Lee, B.; Park, S. Characterization of the complete chloroplast genome of Arabis stellaris and comparisons with related species. PLoS ONE 2017, 12, e0183197. [Google Scholar] [CrossRef]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.M.; Zhao, C.Y.; Liu, X.F. Complete chloroplast genome sequences of Kaempferia galanga and Kaempferia elegans: Molecular structures and comparative analysis. Molecules 2019, 24, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium × hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Nati. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.Y.; Zhao, Y.L.; Xu, Z.G.; Yang, G.Y.; Peng, J.; Peng, X.Y. Initial characterization of the chloroplast genome of Vicia sepium, an important wild resource plant, and related inferences about its evolution. Front. Genet. 2020, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.B.; Yang, S.X.; Li, H.T.; Yang, J.; Li, D.Z. Comparative chloroplast genomes of Camellia species. PLoS ONE 2013, 8, e73053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.L.; Cheng, F.; Rohlsen, D.; Bi, C.W.; Wang, C.Y.; Xu, Y.Q.; Wei, S.Y.; Ye, Q.L.; Yin, T.M.; Ye, N. Organellar genome assembly methods and comparative analysis of horticultural plants. Hortic. Res. 2018, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, M.; Zhu, C.J.; Yang, J.B.; Vatanparast, M.; Schley, R.; Lai, Q.; Zhang, D.Y.; Tu, T.Y.; Klitgård, B.B.; Li, S.J.; et al. Comparative analysis of complete plastid genome reveals powerful barcode regions for identifying wood of Dalbergia odorifera and D. tonkinensis (Leguminosae). J. Syst. Evol. 2020, 60, 73–84. [Google Scholar] [CrossRef]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Sugiura, M.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; Ohto, C.; et al. The complete nucleotide sequence of the Tobacco chloroplast genome: Its gene organization and expression. Plant Mol. Biol. Rep. 1986, 4, 111–148. [Google Scholar] [CrossRef]
- Guo, X.Y.; Liu, J.Q.; Hao, G.Q.; Zhang, L.; Mao, K.S.; Wang, X.J.; Zhang, D.; Ma, T.; Hu, Q.J.; AI-Shehbaz, I.A.; et al. Plastome phylogeny and early diversification of Brassicaceae. BMC Genom. 2017, 18, 176. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.P.; Liu, J.; Jing, Y.; Wang, L.; Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef]
- Dong, W.P.; Liu, H.; Xu, C.; Zuo, Y.J.; Chen, Z.J.; Zhou, S.L. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 138. [Google Scholar] [CrossRef] [Green Version]
- Ni, L.H.; Zhao, Z.L.; Xua, H.X.; Chen, S.L.; Dorje, G. The complete chloroplast genome of Gentiana straminea (Gentianaceae), an endemic species to the Sino-Himalayan subregion. Gene 2016, 577, 281–288. [Google Scholar] [CrossRef]
- Kim, Y.; Cullis, C. A novel inversion in the chloroplast genome of marama (Tylosema esculentum). J. Exp. Bot. 2017, 68, 2065–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Pellizzer, T.; Uibricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Zufall, R.A. Beyond simple homology searches: Multiple sequence alignments and phylogenetic trees. Curr. Protoc. Essent. Lab. Technol. 2009, 1, 11–13. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozas, J.; Sanchez-Delbarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.; Haeseler, A.V.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Haeseler, A.V.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Mark, P.V.D.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, S.; Su, C.; Harris, A.J.; Zhao, L.; Su, N.; Wang, J.R.; Duan, L.; Chang, Z.Y. Comparative chloroplast genomics and phylogenetic analysis of Zygophyllum (Zygophyllaceae) of China. Front. Plant. Sci. 2021, 12, 723622. [Google Scholar] [CrossRef]
- Fu, G.; Liu, J.; Li, J.Q. The complete chloroplast genome sequence of Elsholtzia densa, a herb with volatile aroma component. Mitochondrial DNA B 2020, 5, 595–596. [Google Scholar] [CrossRef] [Green Version]
- Welch, A.J.; Collins, K.; Ratan, A.; Drautz, M.D.I.; Schuster, S.C.; Lindqvist, C. The quest to resolve recent radiations: Plastid phylogenomics of extinct and endangered Hawaiian endemic mints (Lamiaceae). Mol. Phylogenet. Evol. 2016, 99, 16–33. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zhao, Z.Y.; Zhang, T.; Zhong, W.H.; Liu, C.S.; Yuan, Q.J.; Huang, L.Q. The chloroplast genome sequence of Scutellaria baicalensis provides insight into intraspecific and interspecific chloroplast genome diversity in Scutellaria. Genes 2017, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chen, Y.; Lv, J.Z.; Xu, J.; Zhu, S.F.; Li, M.F. Comparative chloroplast genomes of Sorghum species: Sequence divergence and phylogenetic relationships. Biomed. Res. Int. 2019, 19, 5046958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, B.L.H.; Park, H.S.; Lau, T.W.D.; Lin, Z.X.; Yang, J.T.; Shaw, P.C. Comparative analysis and phylogenetic investigation of Hong Kong Ilex chloroplast genomes. Sci. Rep. 2021, 11, 5153. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.L.; Cheng, L.L.; Huang, W.G.; Cao, Q.C.; Zhou, L.; Jia, W.S.; Lan, Y.P. Chloroplast genomes of seven species of Coryloideae (Betulaceae): Structures and comparative analysis. Genome 2020, 63, 337–348. [Google Scholar] [CrossRef]
- Huang, R.; Xie, X.; Li, F.; Tian, E.W.; Chao, Z. Chloroplast genomes of two Mediterranean Bupleurum species and the phylogenetic relationship inferred from combined analysis with East Asian species. Planta 2021, 253, 81. [Google Scholar] [CrossRef]
- Wei, F.; Tang, D.F.; Wei, K.H.; Qin, F.; Li, L.X.; Lin, Y.; Zhu, Y.X.; Khan, A.; Kashif, M.H.; Miao, J.H. The complete chloroplast genome sequence of the medicinal plant Sophora tonkinensis. Sci. Rep. 2020, 10, 12473. [Google Scholar] [CrossRef]
- Curci, P.L.; Demenico, P.D.; Danzi, D.; Vendramin, G.G.; Sonnante, G. Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PLoS ONE 2015, 10, e0120589. [Google Scholar] [CrossRef]
- Wang, K.Y.; Li, L.; Hua, Y.; Zhao, M.Z.; Li, S.K.; Sun, H.H.; Lv, Y.X.; Wang, Y. The complete chloroplast genome of Mentha spicata, an endangered species native to South Europe. Mitochondrial DNA B 2017, 2, 907–909. [Google Scholar] [CrossRef] [Green Version]
- He, Y.H.; Han, L.M.; Liu, Y.P.; Tian, N.; Su, X.; Wang, Z.Z. Complete sequence analysis of chloroplast genome of Salvia japonica. Bull. Bot. Res. 2017, 37, 572–578. [Google Scholar] [CrossRef]
- Hildebrand, M.; Hallick, R.B.; Passavant, C.W.; Bourque, D.P. Trans-splicing in chloroplasts: The rps12 loci of Nicotiana tabacum. Proc. Natl. Acad. Sci. USA 1988, 85, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.C.; Li, Q.S.; Li, Y.; Qian, J.; Han, J.P. Chloroplast genome of Aconitum barbatum var puberulum (Ranunculaceae) derived from CCS reads using the PacBio RS platform. Front. Plant Sci. 2015, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.K.; Cai, Z.Q.; Raubeson, L.A.; Daniell, H.; de Pamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansenet, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, G.; and Wu, W.R. Investigation and utilization of intron length polymorphisms in conifers. New For. 2011, 41, 379–388. [Google Scholar] [CrossRef]
- Ebert, D.; Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cp SSR discovery and applications to a wide array of plant. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef] [PubMed]
- George, B.J.; Bhatt, B.S.; Awasthi, M.; George, B.; Singh, A.K. Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr. Genet. 2015, 61, 665–677. [Google Scholar] [CrossRef]
- Nie, X.J.; Lv, S.Z.; Zhang, Y.X.; Du, X.H.; Wang, L.; Biradar, S.S.; Tan, X.F.; Wan, F.H.; Weining, S. Complete chloroplast genome sequence of a major invasive species, Crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.G.; Zheng, K.X.; Jiao, K.L.; Cai, Y.C.; Chen, C.L.; Mao, Y.Y.; Wang, L.Y.; Zhan, X.R.; Ying, Q.C.; Wang, H.Z. Complete chloroplast genomes of four Physalis species (Solanaceae): Lights into genome structure, comparative analysis, and phylogenetic relationships. BMC Plant Biol. 2020, 20, 242. [Google Scholar] [CrossRef]
- Romero, H.; Zavala, A.; Musto, H. Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic Acids Res. 2000, 28, 2084–2090. [Google Scholar] [CrossRef] [Green Version]
- Morton, B.R. Selection on the codon bias of chloroplast and cyanelle genes in different plant and algal lineages. J. Mol. Evol. 1998, 46, 449–459. [Google Scholar] [CrossRef]
- Qian, J.; Song, J.Y.; Gao, H.H.; Zhu, Y.J.; Xu, J.; Pang, X.H.; Yao, H.; Sun, C.; Li, X.E.; Li, C.Y.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef]
- Yang, L.F.; Yang, Z.Y.; Liu, C.K.; He, Z.S.; Zhang, Z.R.; Yang, J.; Liu, H.Y.; Yang, J.B.; Ji, Y.H. Chloroplast phylogenomic analysis provides insights into the evolution of the largest eukaryotic genome holder, Paris japonica (Melanthiaceae). BMC Plant Biol. 2019, 19, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulding, S.E.; Olmstead, R.G.; Morden, C.W.; Wolfe, K.H. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 1996, 252, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Gao, H.H.; Wang, Y.T.; Song, J.Y.; Robert, H.; Wu, H.Z.; Hu, Z.G.; Yao, H.; Luo, H.M.; Luo, K.; et al. Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Sci. China Life Sci. 2013, 56, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Alzahrani, D.A.; Yaradua, S.S.; Albokhari, E.J.; Abba, A. Complete chloroplast genome sequence of Barleria prionitis, comparative chloroplast genomics and phylogenetic relationships among Acanthoideae. BMC Genom. 2020, 21, 393. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Xu, C.; Li, C.H.; Sun, J.H.; Zuo, Y.J.; Shi, S.; Cheng, T.; Guo, J.J.; Zhou, S.L. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, J.; Rousseau-Gueutin, M.; Martin, G.E.; Morice, J.; Boutte, J.; Coissac, E.; Ourari, M.; Aïnouche, M.; Salmon, A.; Cabello-Hurtado, F.; et al. The evolutionary fate of the chloroplast and nuclear rps16 genes as revealed through the sequencing and comparative analyses of four novel legume chloroplast genomes from Lupinus. DNA Res. 2017, 24, 343–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Nielsen, R. Estimating synonymous and non-synonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Makalowski, W.; Boguski, M.S. Evolutionary parameters of the transcribed mammalian genome: An analysis of 2820 orthologous rodent and human sequences. Proc. Natl. Acad. Sci. USA 1998, 95, 9407–9412. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.H.; Liao, R.; Yang, T.G.; Dong, X.; Lan, D.Q.; Qin, R.; Liu, H. Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genom. 2020, 21, 621. [Google Scholar] [CrossRef]
- Zhao, F.; Chen, Y.P.; Salmaki, Y.; Drew, B.T.; Wilson, T.; Scheen, A.C.; Celep, F.; Bräuchler, C.; Bendiksby, M.; Wang, Q.; et al. An updated tribal classification of Lamiaceae based on plastome phylogenomics. BMC Biol. 2021, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.C.; Bendiksby, M.; Ryding, O.; Mathiesen, C.; Albert, V.A.; Lindqvist, C. Molecular phylogenetics, character evolution and suprageneric classification of Lamioideae (Lamiaceae). Ann. Mo. Bot. Gard. 2010, 97, 191–217. [Google Scholar] [CrossRef]
- Bendiksby, M.; Thorbek, L.; Scheen, A.C.; Lindqvist, C.; Ryding, O. An updated phylogeny and classification of Lamiaceae subfamily Lamioideae. Taxon 2011, 60, 471–484. [Google Scholar] [CrossRef]
- Burke, S.V.; Lin, C.S.; Wysocki, W.P.; Clark, L.G.; Duvall, M.R. Phylogenomics and plastome evolution of tropical forest grasses (Leptaspis, Streptochaeta: Poaceae). Front. Plant Sci. 2016, 7, 1993. [Google Scholar] [CrossRef] [Green Version]
- Wagstaff, S.J.; Olmstead, R.G.; Cantino, P.D. Parsimony analysis of cpDNA restriction site variation in subfamily Nepetoideae (Labiatae). Am. J. Bot. 1995, 82, 886–892. [Google Scholar] [CrossRef]
- Wagstaff, S.J.; Hickerson, L.; Spangler, R.; Reeves, P.A.; Olmstead, R.G. Phylogeny in Labiatae s.l., inferred from cpDNA sequences. Pl. Syst. Evol. 1998, 209, 265–274. [Google Scholar] [CrossRef]
- Paton, A.J.; Springate, D.; Suddee, S.; Otieno, D.; Grayer, R.J.; Harley, M.M.; Willis, F.; Simmonds, M.S.J.; Powell, M.P.; Savolainenb, V. Phylogeny and evolution of basils and allies (Ocimeae, Labiatae) based on three plastid DNA regions. Mol. Phylogenet. Evol. 2004, 31, 277–299. [Google Scholar] [CrossRef]
- Drew, B.T.; Sytsma, K.J. Phylogenetics, biogeography, and staminal evolution in the tribe Mentheae (Lamiaceae). Am. J. Bot. 2012, 99, 933–953. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Qi, Z.C.; Liu, L.X.; Ohi-Toma, T.; Lee, J.; Hsieh, T.H.; Fu, C.X.; Cameron, K.M.; Qiu, Y.X. Molecular phylogenetics and biogeography of the mint tribe Elsholtzieae (Nepetoideae, Lamiaceae), with an emphasis on its diversification in East Asia. Sci. Rep. 2017, 7, 2057. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.P.; Drew, B.T.; Li, B.; Soltis, D.E.; Soltis, P.S.; Xiang, C.L. Resolving the phylogenetic position of Ombrocharis (Lamiaceae), with reference to the molecular phylogeny of tribe Elsholtzieae. Taxon 2016, 65, 123–136. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | D. heterophyllum | D. tanguticum | D. moldavica | D. palmatum | D. taliense |
|---|---|---|---|---|---|
| GenBank accession | OM201748 | MT457746 | MT457747 | NC_031874.1 | MT473756 |
| Size (bp) | 150,869 | 150,594 | 149,868 | 150,510 | 150,976 |
| GC content (%) | 37.8 | 37.8 | 37.8 | 37.8 | 37.8 |
| IR (bp) | 51,350 | 51,370 | 51,352 | 50,772 | 51,358 |
| GC content in IR (%) | 43.2 | 43.0 | 43.0 | 43.1 | 43.0 |
| LSC (bp) | 82,421 | 82,221 | 81,450 | 82,160 | 82,253 |
| GC content in LSC (%) | 35.7 | 35.8 | 35.8 | 35.8 | 35.8 |
| SSC (bp) | 17,098 | 17,363 | 17,066 | 17,254 | 17,365 |
| GC content in SSC (%) | 31.8 | 31.9 | 31.8 | 31.8 | 31.6 |
| Gene number | 133 | 133 | 132 | 125 | 134 |
| tRNA gene number | 37 | 37 | 37 | 29 | 37 |
| rRNA gene number | 8 | 8 | 8 | 8 | 8 |
| Protein-coding gene | 88 | 88 | 87 | 88 | 89 |
| Category | Group of Gene | Gene Name |
|---|---|---|
| Gene for photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of NADH dehydrogenase | ndhA, ndhB1 (2×), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB1, petD1, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF1, atpH, atpI | |
| Large subunit of rubisco | rbcL | |
| Self-replication | Proteins of large ribosomal subunit | rpl21 (2×), rpl14, rpl161, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 |
| Proteins of small ribosomal subunit | rps2, rps3, rps4, rps7 (2×), rps8, rps11, rps12 (2×), rps14, rps15, rps16 1, rps18, rps19 | |
| Subunits of RNA polymerase Ribosomal RNA | rrn4.5 (2×), rrn5 (2×), rrn16 (2×), rrn23 (2×) | |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC11, rpoC2 | |
| Transfer RNA | trnA-UGC1 (2×), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnG-UCC1, trnH-GUG, trnI-CAU (2×), trnI-GAU 1 (2×), trnK-UUU1, trnL-CAA (2×), trnL-UAG, trnL-UAA1, trnfM-CAU, trnM-CAU, trnN-GUU (2×), trnP-UGG, trnQ-UUG, trnR-ACG (2×), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC (2×), trnV-UAC 1, trnW-CCA, trnY-GUA | |
| Biosynthesis | Maturase | matK |
| Protease | clpP2 | |
| Envelope membrane protein | cemA | |
| Subunit Acetyl-CoA carboxylase | accD | |
| C-type cytochrome synthesis gene | ccsA | |
| Translation initiation factor | infA | |
| Unknown | Conserved open reading frame | ycf1 (2×), ycf2 (2×), ycf3 2, ycf4, ycf15 (2×) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, G.; Liu, Y.; Caraballo-Ortiz, M.A.; Zheng, C.; Liu, T.; Xu, Y.; Su, X. Characterization of the Complete Chloroplast Genome of the Dragonhead Herb, Dracocephalumheterophyllum (Lamiaceae), and Comparative Analyses with Related Species. Diversity 2022, 14, 110. https://doi.org/10.3390/d14020110
Fu G, Liu Y, Caraballo-Ortiz MA, Zheng C, Liu T, Xu Y, Su X. Characterization of the Complete Chloroplast Genome of the Dragonhead Herb, Dracocephalumheterophyllum (Lamiaceae), and Comparative Analyses with Related Species. Diversity. 2022; 14(2):110. https://doi.org/10.3390/d14020110
Chicago/Turabian StyleFu, Gui, Yuping Liu, Marcos A. Caraballo-Ortiz, Changyuan Zheng, Tao Liu, Yujie Xu, and Xu Su. 2022. "Characterization of the Complete Chloroplast Genome of the Dragonhead Herb, Dracocephalumheterophyllum (Lamiaceae), and Comparative Analyses with Related Species" Diversity 14, no. 2: 110. https://doi.org/10.3390/d14020110
APA StyleFu, G., Liu, Y., Caraballo-Ortiz, M. A., Zheng, C., Liu, T., Xu, Y., & Su, X. (2022). Characterization of the Complete Chloroplast Genome of the Dragonhead Herb, Dracocephalumheterophyllum (Lamiaceae), and Comparative Analyses with Related Species. Diversity, 14(2), 110. https://doi.org/10.3390/d14020110