
Chloroplast Genome of Salvia Sect. Drymosphace: Comparative and Phylogenetic Analysis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, DNA Extraction and Genome Sequencing
2.2. Plastome Assembly and Annotation
2.3. Codon Usage and Repeated Sequence Analysis
2.4. Comparative Genomic Analyses
2.5. Phylogenetic Analysis
3. Results and Discussion
3.1. Plastome Features of Sect. Drymosphace
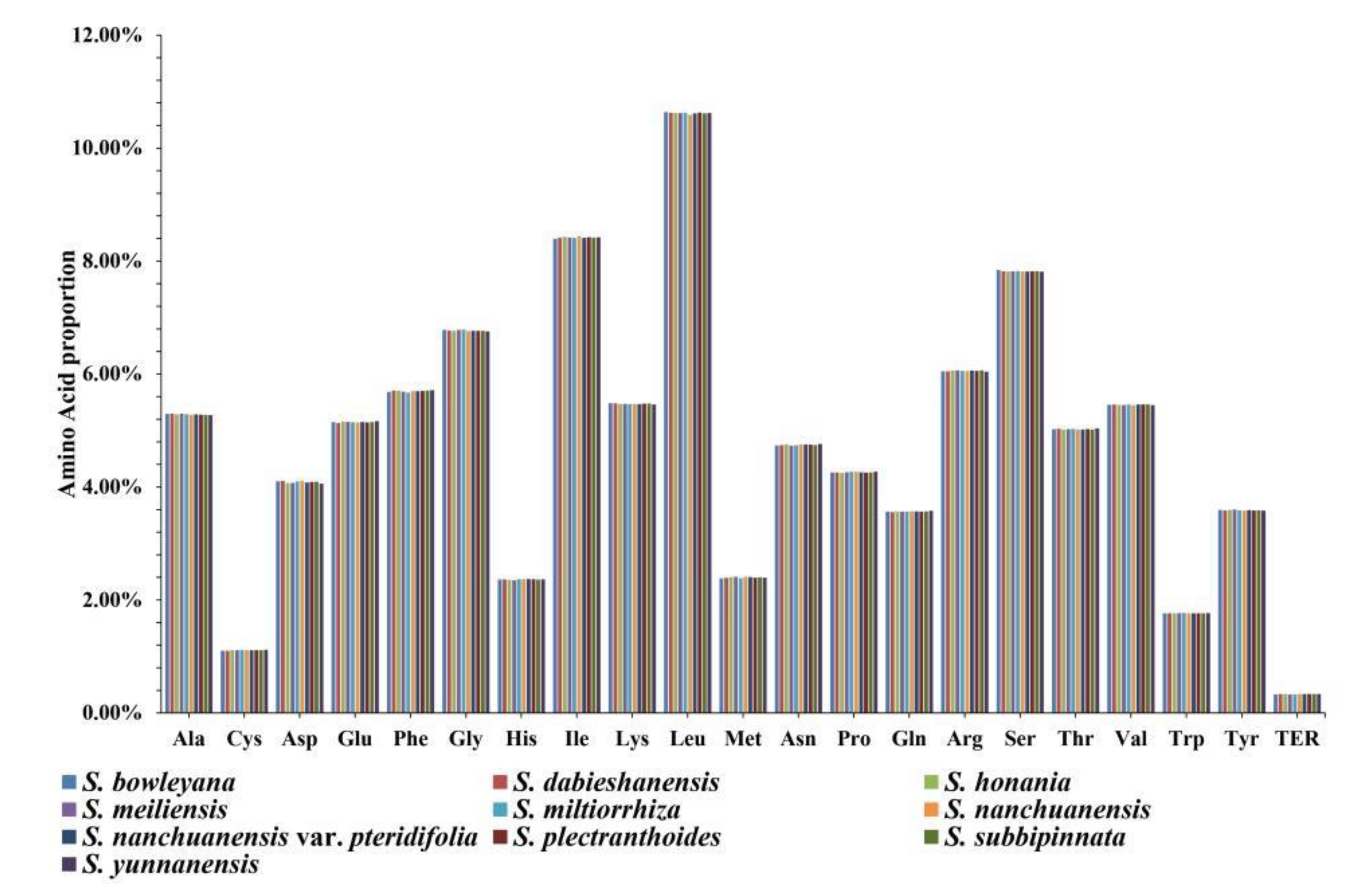
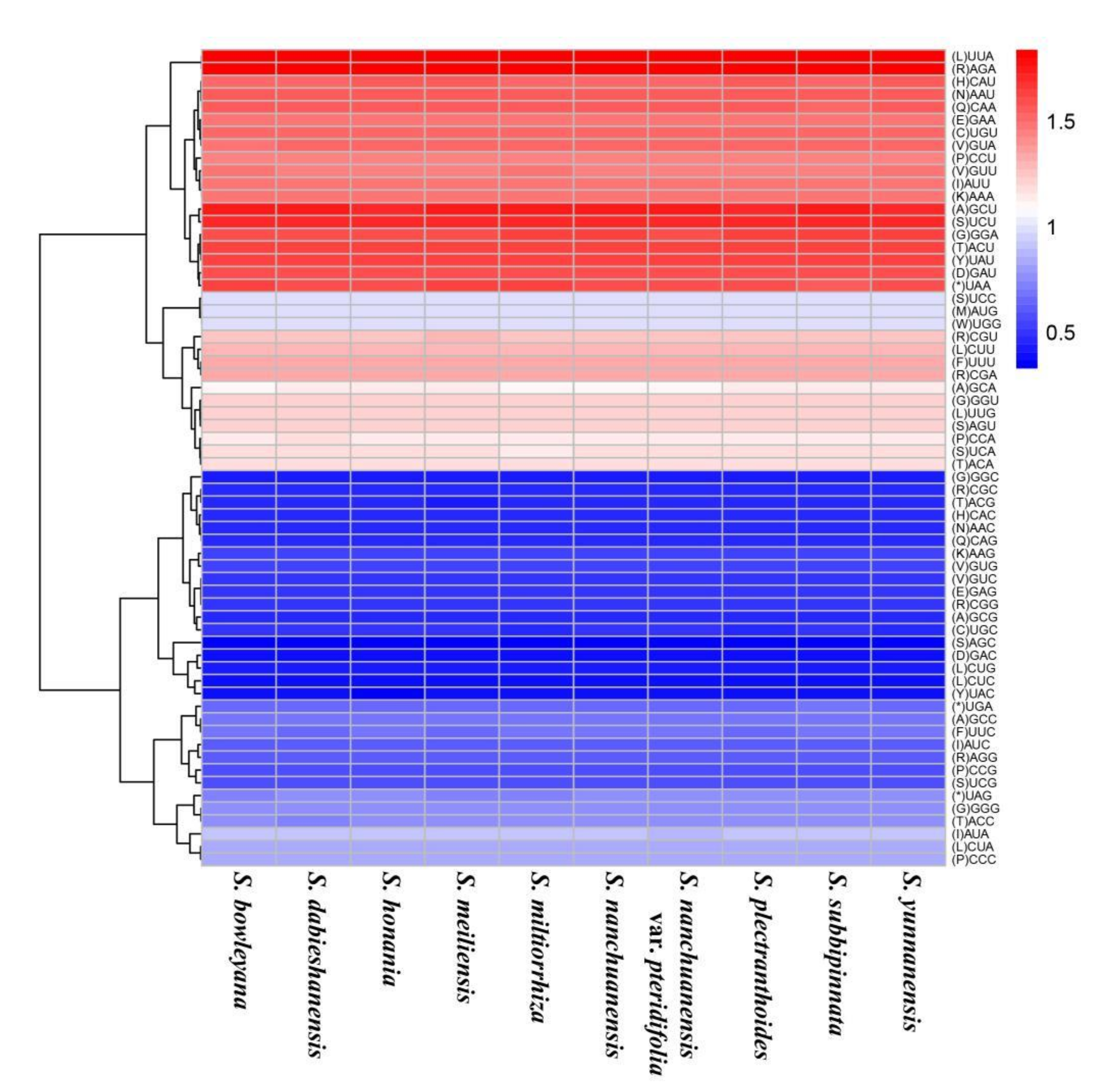
3.2. Amino Acid Abundance and Codon Usage
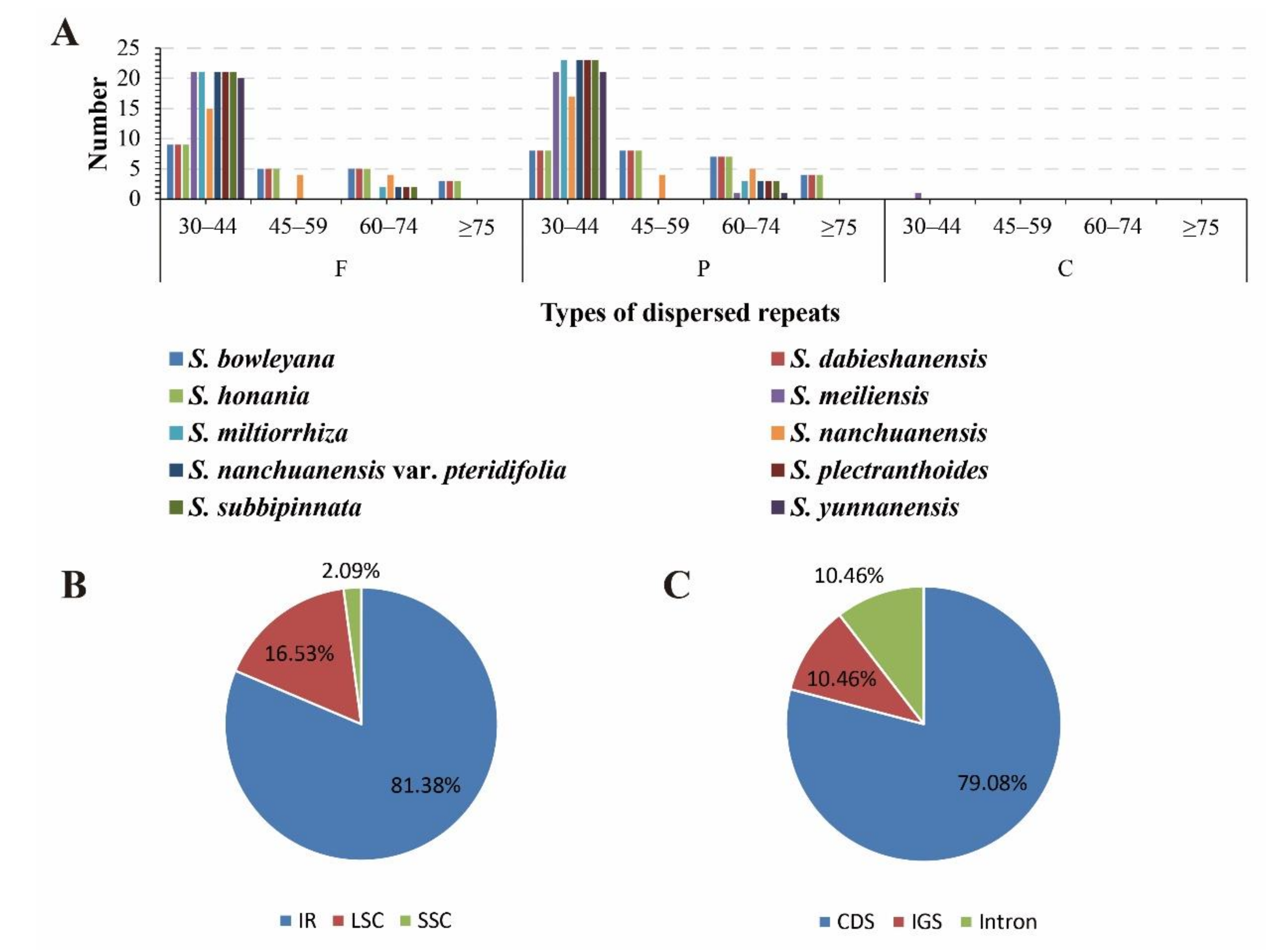
3.3. Simple Sequence Repeats and Long Repeat Sequences
3.4. Comparative Genomic Analyses
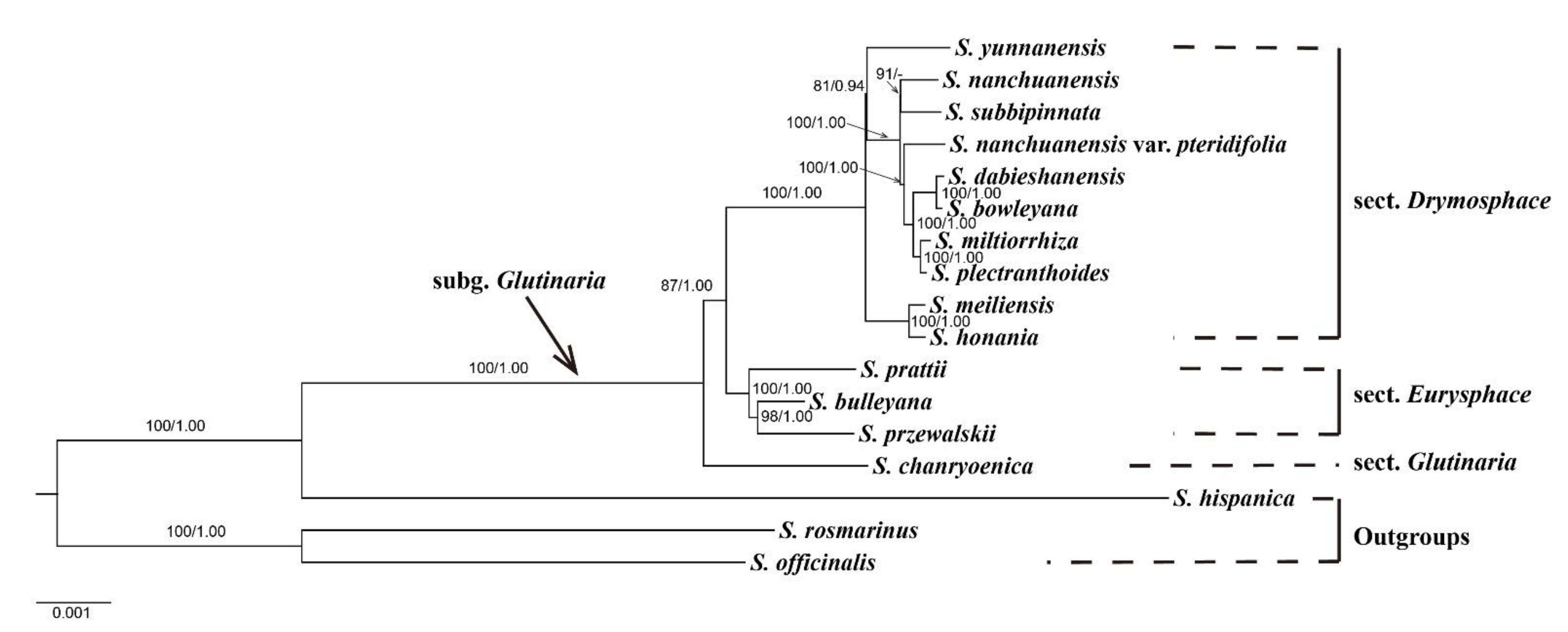
3.5. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drew, B.; González-Gallegos, J.G.; Xiang, C.L.; Kriebel, R.; Drummond, C.; Walker, J.; Sytsma, K. Salvia united: The greatest good for the greatest number. Taxon 2017, 66, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.X.; Takano, A.; Drew, B.T.; Liu, E.D.; Soltis, D.E.; Soltis, P.S.; Peng, H.; Xiang, C.L. Phylogeny and staminal evolution of Salvia (Lamiaceae, Nepetoideae) in East Asia. Ann. Bot. 2018, 122, 649–668. [Google Scholar] [CrossRef] [PubMed]
- Kriebel, R.; Drew, B.T.; Drummond, C.P.; Gonzalez-Gallegos, J.G.; Celep, F.; Mahdjoub, M.M.; Rose, J.P.; Xiang, C.L.; Hu, G.X.; Walker, J.B.; et al. Tracking temporal shifts in area, biomes, and pollinators in the radiation of Salvia (sages) across continents: Leveraging anchored hybrid enrichment and targeted sequence data. Am. J. Bot. 2019, 106, 573–597. [Google Scholar] [CrossRef] [Green Version]
- Li, H.W.; Hedge, I.C. Salvia. In Flora of China; Wu, C.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 1994; Volume 17, pp. 196–224. [Google Scholar]
- Hu, G.X.; Liu, Y.; Xu, W.B.; Liu, E.D. Salvia petrophila sp. nov. (Lamiaceae) from north Guangxi and south Guizhou, China. Nord. J. Bot. 2014, 32, 190–195. [Google Scholar] [CrossRef]
- Hu, G.X.; Liu, E.D.; Zhang, T.; Cai, J.; Xiang, C.L. Salvia luteistriata (Lamiaceae), a new species from northeastern Sichuan, China. Phytotaxa 2017, 314, 123–128. [Google Scholar] [CrossRef]
- Hu, G.X.; Peng, H. Identity of Salvia weihaiensis (Lamiaceae) from China. Phytotaxa 2015, 202, 298–300. [Google Scholar] [CrossRef]
- Xiang, C.L.; Hu, G.X.; Peng, H. New combinations and new synonyms in Lamiaceae from China. Biodivers. Sci. 2016, 24, 719–722. [Google Scholar] [CrossRef]
- Ding, B.Y.; Chen, Z.H.; Xu, Y.L.; Jin, X.F.; Wu, D.F.; Chen, J.B.; Wu, W.J. New species and combination of Lamiaceae from Zhejiang China. Guihaia 2019, 39, 10–15. [Google Scholar] [CrossRef]
- Wei, Y.K.; Pendry, C.A.; Zhang, D.G.; Huang, Y.B. Salvia daiguii (Lamiaceae): A new species from west Hunan, China. Edinb. J. Bot. 2019, 76, 359–368. [Google Scholar] [CrossRef]
- Wei, Y.K.; Pendry, C.A.; Huang, Y.B.; Ge, B.J.; Xiao, H.W. Salvia subviolacea, A new species from the Himalayas-Hengduan Mountains, China. Edinb. J. Bot. 2021, 78, 1–9. [Google Scholar] [CrossRef]
- Hu, G.X.; Liu, E.D.; Wu, Z.K.; Sytsma, K.J.; Drew, B.T.; Xiang, C.L. Integrating DNA sequences with morphological analysis clarifies phylogenetic position of Salvia grandifolia (Lamiaceae): An enigmatic species endemic to Southwestern China. Int. J. Plant Sci. 2020, 181, 787–799. [Google Scholar] [CrossRef]
- Bentham, G. Labiatarum Genera Et Species; Ridgeway: London, UK, 1932–1936. [Google Scholar]
- Bentham, G. Labiatae. In Genera Plantarum; Bentham, G., Hooker, J.D., Eds.; Reeve and Co.: London, UK, 1876; Volume 2, pp. 1160–1196. [Google Scholar]
- Stibal, E. A revision of Chinese and East Burmese species of Salvia. Acta Horti Gotobg. 1934, 9, 101–165. [Google Scholar]
- Wu, C.Y. Salvia. In Flora Reipublicae Popularis Sinicae; Wu, C.Y., Ed.; Science Press: Beijing, China, 1977; Volume 66, pp. 70–196. [Google Scholar]
- Wang, B.Q. Salvia miltiorrhiza: Chemical and pharmacological review of a medicinal plant. J. Med. Plant. Res. 2010, 4, 2813–2820. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.B.; Sytsma, K.J. Staminal evolution in the genus Salvia (Lamiaceae): Molecular phylogenetic evidence for multiple origins of the staminal lever. Ann. Bot. 2007, 100, 375–391. [Google Scholar] [CrossRef]
- Jenks, A.A.; Walker, J.B.; Kim, S.C. Phylogeny of New World Salvia subgenus Calosphace (Lamiaceae) based on cpDNA (psbA-trnH) and nrDNA (ITS) sequence data. J. Plant Res. 2013, 126, 483–496. [Google Scholar] [CrossRef]
- Will, M.; Classen-Bockhoff, R. Why Africa matters: Evolution of Old World Salvia (Lamiaceae) in Africa. Ann. Bot. 2014, 114, 61–83. [Google Scholar] [CrossRef] [Green Version]
- Will, M.; Classen-Bockhoff, R. Time to split Salvia s.l. (Lamiaceae)-New insights from Old World Salvia phylogeny. Mol. Phylogenet. Evol. 2017, 109, 33–58. [Google Scholar] [CrossRef]
- Fragoso-Martínez, I.; Martínez-Gordillo, M.; Salazar, G.A.; Sazatornil, F.; Jenks, A.A.; Peña, M.R.G.; Barrera-Aveleida, G.; Benitez-Vieyra, S.; Magallón, S.; Cornejo-Tenorio, G. Phylogeny of the Neotropical sages (Salvia subg. Calosphace; Lamiaceae) and insights into pollinator and area shifts. Plant Syst. Evol. 2018, 304, 43–55. [Google Scholar] [CrossRef]
- Deng, T.; Nie, Z.L.; Drew, B.T.; Volis, S.; Kim, C.; Xiang, C.L.; Zhang, J.W.; Wang, Y.H.; Sun, H. Does the Arcto-Tertiary biogeographic hypothesis explain the disjunct distribution of Northern Hemisphere herbaceous plants? The case of Meehania (Lamiaceae). PLoS ONE 2015, 10, e0117171. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [Green Version]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Li, B.; Drew, B.T.; Chen, Y.P.; Wang, Q.; Yu, W.B.; Liu, E.D.; Salmaki, Y.; Peng, H.; Xiang, C.L. Leveraging plastomes for comparative analysis and phylogenomic inference within Scutellarioideae (Lamiaceae). PLoS ONE 2020, 15, e0232602. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ma, P.F.; Li, H.T.; Hu, G.X.; Li, D.Z. Comparative plastomic analysis and insights into the phylogeny of Salvia (Lamiaceae). Plant Divers. 2021, 43, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Drew, B.T.; Chen, Y.P.; Hu, G.X.; Li, B.; Xiang, C.L. The Chloroplast genome of Salvia: Genomic characterization and phylogenetic analysis. Int. J. Plant Sci. 2020, 181, 812–830. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; De Pamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhang, J.; Yuan, G.; Liu, C. Complex interplay among DNA modification, noncoding RNA expression and protein-coding RNA expression in Salvia miltiorrhiza chloroplast genome. PLoS ONE 2014, 9, e99314. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 1999. [Google Scholar]
- Randall, T.A.; Dwyer, R.A.; Huitema, E.; Beyer, K.; Cvitanich, C.; Kelkar, H.; Fong, A.M.; Gates, K.; Roberts, S.; Yatzkan, E.; et al. Large-scale gene discovery in the Oomycete Phytophthora infestans reveals likely components of phytopathogenicity shared with true fungi. Mol. Plant Microbe Interact. 2005, 18, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; IEEE: Manhattan, NY, USA, 2010. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Gao, F.L.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef]
- Gao, B.; Yuan, L.; Tang, T.; Hou, J.; Pan, K.; Wei, N. The complete chloroplast genome sequence of Alpinia oxyphylla Miq. and comparison analysis within the Zingiberaceae family. PLoS ONE 2019, 14, e0218817. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Zhou, A.; Zhang, Y.; Zou, X.; Li, D.; Duan, A.; He, C. Characterization of the complete chloroplast genomes of five Populus species from the western Sichuan plateau, southwest China: Comparative and phylogenetic analyses. PeerJ 2019, 7, e6386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.X.; Hu, G.X.; Hu, G.W. Comparative genomics and phylogenetic relationships of two endemic and endangered species (Handeliodendron bodinieri and Eurycorymbus cavaleriei) of two monotypic genera within Sapindales. BMC Genom. 2022, 23, 27. [Google Scholar] [CrossRef]
- Gu, L.; Su, T.; An, M.T.; Hu, G.X. The Complete dhloroplast genome of the vulnerable Oreocharis esquirolii (Gesneriaceae): Structural features, comparative and phylogenetic Analysis. Plants 2020, 9, 1692. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef]
- Shimada, H.; Sugiura, M. Fine structural features of the chloroplast genome: Comparison of the sequenced chloroplast genomes. Nucleic Acids Res. 1991, 19, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Kim, W.J.; Yeo, S.M.; Choi, G.; Kang, Y.M.; Piao, R.; Moon, B.C. The complete chloroplast genome sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and comparative analysis with Other Fritillaria species. Molecules 2017, 22, 982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Zhou, P.; Choi, Y.A.; Huang, S.; Gmitter, F.G., Jr. Mining and characterizing microsatellites from Citrus ESTs. Theor. Appl. Genet. 2006, 112, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; He, R.; Lu, J.; Jiang, M.; Shen, X.; Jiang, Y.; Wang, Z.; Wang, H. Development of SSR markers and assessment of genetic diversity in medicinal Chrysanthemum morifolium cultivars. Front. Genet. 2016, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xiao, H.; Deng, C.; Xiong, L.; Yang, J.; Peng, C. The complete chloroplast genome sequences of the medicinalplant Pogostemon cablin. Int. J. Mol Sci. 2016, 17, 820. [Google Scholar] [CrossRef] [Green Version]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.P.; Bi, Y.; Yang, F.P.; Zhang, M.F.; Chen, X.Q.; Xue, J.; Zhang, X.H. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.L.; Wang, L.; Lei, J.; Duan, B.Z.; Ma, W.S.; Xiao, S.M.; Qi, H.J.; Wang, Z.; Liu, Y.Q.; Shen, X.F.; et al. A comparative analysis of the chloroplast genomes of four Salvia medicinal plants. Engineering 2019, 5, 907–915. [Google Scholar] [CrossRef]
- Liang, C.L.; Wang, L.; Ma, W.S.; Xu, J. A comparative study of complete chloroplast genome for the genus Salvia. J. Plant Biochem. Biot. 2021, 30, 117–125. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 2002, 12, R62–R64. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.I.; Soreng, R.J. Migration of endpoints of two genes relative to boundaries between regions of the plastid genome in the grass family (Poaceae). Am. J. Bot. 2010, 97, 874–892. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Ma, P.F.; Wen, J.; Yi, T.S. Complete sequencing of five Araliaceae chloroplast genomes and the phylogenetic implications. PLoS ONE 2013, 8, e78568. [Google Scholar] [CrossRef]
- Menezes, A.P.A.; Resende-Moreira, L.C.; Buzatti, R.S.O.; Nazareno, A.G.; Carlsen, M.; Lobo, F.P.; Kalapothakis, E.; Lovato, M.B. Chloroplast genomes of Byrsonima species (Malpighiaceae): Comparative analysis and screening of high divergence sequences. Sci. Rep. 2018, 8, 2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Yang, M.; Mo, C.; Xie, W.; Liu, C.; Wu, B.; Ma, X. Complete chloroplast genomes of two Siraitia Merrill species: Comparative analysis, positive selection and novel molecular marker development. PLoS ONE 2019, 14, e0226865. [Google Scholar] [CrossRef]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef]
- Meng, X.X.; Xian, Y.F.; Xiang, L.; Zhang, D.; Shi, Y.H.; Wu, M.L.; Dong, G.Q.; Ip, S.P.; Lin, Z.X.; Wu, L.; et al. Complete chloroplast genomes from Sanguisorba: Identity and variation among four species. Molecules 2018, 23, 2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, W.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, W.J.; Erickson, D.L. A two-locus global DNA barcode for land plants: The coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS ONE 2007, 2, e508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, M.W.; Cowan, R.S.; Hollingsworth, P.M.; Berg, C.V.D.; Wilkinson, M.J. A Proposal for a Standardised Protocol to Barcode All Land Plants. Taxon 2007, 56, 295–299. [Google Scholar] [CrossRef]
- Group, C.P.W. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar]
- Meng, H.H.; Zhang, M.L. Phylogeography of Lagochilus ilicifolius (Lamiaceae) in relation to Quaternary climatic oscillation and aridification in northern China. Biochem. Syst. Ecol. 2021, 39, 787–796. [Google Scholar] [CrossRef]
- Hu, G.X.; Su, T.; An, M.T.; Wang, X.Y. Rediscovery of Pogostemon dielsianus (Lamiaceae, Lamioideae), a rare endemic species from southwestern China, after one century. PhytoKeys 2021, 171, 61–73. [Google Scholar] [CrossRef]
- Xiang, C.L.; Zhao, F.; Cantino, P.D.; Drew, B.T.; Li, B.; Liu, E.D.; Soltis, D.E.; Soltis, P.S.; Peng, H. Molecular systematics of Caryopteris (Lamiaceae) and its allies with reference to the molecular phylogeny of subfamily Ajugoideae. Taxon 2018, 67, 376–394. [Google Scholar] [CrossRef]
- Li, Q.Q.; Li, M.H.; Yuan, Q.J.; Cui, Z.H.; Huang, L.Q.; Xiao, P.G. Phylogenetic relationships of Salvia (Lamiaceae) in China: Evidence from DNA sequence datasets. J. Syst. Evol. 2013, 51, 184–195. [Google Scholar] [CrossRef]
- Drew, B.T.; Sytsma, K.J. Testing the monophyly and placement of Lepechinia in the tribe Mentheae (Lamiaceae). Syst. Bot. 2011, 36, 1038–1049. [Google Scholar] [CrossRef]
- Su, S.W.; Shen, Z.A.; He, J.Q. New species of the genus Salvia from Anhui. Acta Bot. Yunnanica 1984, 6, 55–62. [Google Scholar]
- Rokas, A.; Carroll, S.B. More genes or more taxa? The relative contribution of gene number and taxon number to phylogenetic accuracy. Mol. Biol. Evol. 2005, 22, 1337–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippe, H.; Brinkmann, H.; Lavrov, D.V.; Littlewood, D.T.J.; Manuel, M.; Wörheide, G.; Baurain, D. Resolving difficult phylogenetic questions: Why more sequences are not enough. PLoS Biol. 2011, 9, e1000602. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Subgenus | Voucher | Locality | GenBank Accession | SRA Accession |
|---|---|---|---|---|---|
| S. bowleyana | Glutinaria | GX Hu and F Zhao 131 | China, Fujian | MW435404 * | SRR18650098 * |
| S. bulleyana | Glutinaria | NA | NA | MH603954 | NA |
| S. chanryoenica | Glutinaria | s.n. (KH) | South Korea | MH261357 | NA |
| S. dabieshanensis | Glutinaria | GX Hu and F Zhao 0165 | China, Anhui | MW435405 * | SRR18587923 * |
| S. honania | Glutinaria | GX Hu and F Zhao 0168 | China, Henan | MW435406 * | SRR18600490 * |
| S. meiliensis | Glutinaria | GX Hu and FZ Shangguan Hu0089 | China, Anhui | MN520018 | NA |
| S. miltiorrhiza | Glutinaria | Cultivated | China, Beijing | HF586694 | NA |
| S. nanchuanensis | Glutinaria | JX Yang and XZ He YJX-01 | China, Hunan | MW435407 * | SRR18595850 * |
| S. nanchuanensis var. pteridifolia | Glutinaria | GX Hu and B Pan 615 | China, Guangxi | MW435408 * | SRR18595337 * |
| S. plectranthoides | Glutinaria | GX Hu et al. 0006 | China, Yunnan | MW435409 * | SRR18650097 * |
| S. prattii | Glutinaria | NA | China, Yunnan | MK944407 | NA |
| S. przewalskii | Glutinaria | NA | NA | MH603953 | NA |
| S. subbipinnata | Glutinaria | JX Yang and XZ He YJX-04 | China, Zhejiang | MW435410 * | SRR18650096 * |
| S. yunnanensis | Glutinaria | GX Hu 603 | China, Guizhou | MW435411 * | SRR18595336 * |
| S. hispanica | Calosphace | Cultivated, SD118 | China, Shandong | MT083896 | NA |
| S. rosmarinus | Rosmarinus | Cultivated | China, Shaanxi | KR232566 | NA |
| S. officinalis | Salvia | So_003 | NA | MG772529 | NA |
| Data Matrix | Aligned Length (bp) | Constant Sites (bp) | Variable Sites (bp) | Parsimony Informative (bp) | Best Fit Model (AIC) |
|---|---|---|---|---|---|
| CP | 151,218 | 144,550 (95.59%) | 6668 | 2366 (1.56%) | GTR + F + I + G4 |
| LSC | 82,556 | 77,911 (94.37%) | 4645 | 1637 (1.98%) | GTR + F + I + G4 |
| SSC | 17,531 | 15,967 (91.08%) | 1564 | 567 (3.23%) | GTR + F + G4 |
| IR | 25,563 | 25,335 (99.11%) | 228 | 80 (0.31%) | GTR + F + I |
| CR | 66,888 | 64,304 (96.14%) | 2584 | 921 (1.38%) | GTR + F + I + G4 |
| NCR | 59,877 | 56,165 (93.80%) | 3712 | 1346 (2.25%) | GTR + F + I + G4 |
| HVR | 10,298 | 9096 (88.33%) | 1064 | 363 (3.52%) | GTR + F + G4 |
| Taxa | Clean Reads | Reads Used for Assembly | Mean Coverage (×) | Complete | LSC | SSC | IR | Number of Genes | PCG | tRNA Genes | rRNA Genes | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size(bp) | GC (%) | Length (bp) | GC (%) | Length (bp) | GC (%) | Length (bp) | GC (%) | ||||||||
| S. bowleyana | 6961,322 | 6,160,014 | 526 | 151,508 | 38.01 | 82,809 | 36.14 | 17,585 | 32.01 | 25,557 | 43.12 | 114 | 80 | 30 | 4 |
| S. dabieshanensis | 11,727,732 | 9,644,622 | 504 | 151,330 | 38.02 | 82,629 | 36.14 | 17,587 | 32.01 | 25,557 | 43.12 | 114 | 80 | 30 | 4 |
| S. honania | 7,221,162 | 6,369,236 | 514 | 151,522 | 38.01 | 82,768 | 36.13 | 17,586 | 31.97 | 25,584 | 43.11 | 114 | 80 | 30 | 4 |
| S. meiliensis | 68,112,046 | 25,577,470 | 1970 | 151,614 | 38.00 | 82,854 | 36.12 | 17,580 | 32.00 | 25,590 | 43.10 | 114 | 80 | 30 | 4 |
| S. miltiorrhiza | NA | NA | NA | 151,332 | 38.02 | 82,698 | 36.15 | 17,556 | 32.01 | 25,539 | 43.12 | 114 | 80 | 30 | 4 |
| S. nanchuanensis | 9,188,568 | 7,579,704 | 507 | 151,426 | 38.02 | 82,791 | 36.14 | 17,563 | 31.99 | 25,536 | 43.13 | 114 | 80 | 30 | 4 |
| S. nanchuanensis var. pteridifolia | 15,948,806 | 13,152,169 | 514 | 151,455 | 38.01 | 82,831 | 36.12 | 17,584 | 32.05 | 25,520 | 43.14 | 114 | 80 | 30 | 4 |
| S. plectranthoides | 6,978,570 | 6,231,469 | 512 | 151,416 | 38.01 | 82,775 | 36.13 | 17,563 | 32.03 | 25,539 | 43.12 | 114 | 80 | 30 | 4 |
| S. subbipinnata | 5,368,560 | 4,649,062 | 516 | 151,388 | 38.03 | 82,769 | 36.15 | 17,541 | 32.02 | 25,539 | 43.13 | 114 | 80 | 30 | 4 |
| S. yunnanensis | 6,448,098 | 5,394,701 | 508 | 151,413 | 38.02 | 82,652 | 36.14 | 17,581 | 32.03 | 25,590 | 43.11 | 114 | 80 | 30 | 4 |
| Gene Functions | Group of Genes | Name of Genes |
|---|---|---|
| Photosynthesis | Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI |
| Subunits of NADH dehydrogenase | ndhA *, ndhB * (×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome | petA, petB *, petD *, petG, petL, petN | |
| Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunit of rubisco | rbcL | |
| Self-replication | Large subunit of ribosome | rpl2 * (×2), rpl14, rpl16 *, rpl20, rpl22, rpl23 (×2), rpl32, rpl33, rpl36 |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | |
| Small subunit of ribosome | rps2, rps3, rps4, rps7 (×2), rps8, rps11, rps12 ** (×2), rps14, rps15, rps16*, rps18, rps19 | |
| rRNA Genes | rrn4.5 (×2), rrn5 (×2), rrn16 (×2), rrn23 (×2) | |
| tRNA Genes | trnA-UGC * (×2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC *, trnH-GUG, trnI-CAU (×2), trnI-GAU * (×2), trnK-UUU *, trnL-CAA (×2), trnL-UAA *, trnL-UAG, trnM-CAU, trnN-GUU (×2), trnP-UGG, trnQ-UUG, trnR-ACG (×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC (×2), trnV-UAC *, trnW-CCA, trnY-GUA | |
| Other genes | Subunit of Acetyl-CoA-carboxylase | accD |
| c-type cytochrome synthesis gene | ccsA | |
| Envelop membrane protein | cemA | |
| Protease | clpP ** | |
| Translational initiation | infA | |
| Maturase | matK | |
| Unknown function | Conserved open reading frames | ycf1, ycf2 (×2), ycf3 **, ycf4, ycf15 (×2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, T.; Geng, Y.-F.; Xiang, C.-L.; Zhao, F.; Wang, M.; Gu, L.; Hu, G.-X. Chloroplast Genome of Salvia Sect. Drymosphace: Comparative and Phylogenetic Analysis. Diversity 2022, 14, 324. https://doi.org/10.3390/d14050324
Su T, Geng Y-F, Xiang C-L, Zhao F, Wang M, Gu L, Hu G-X. Chloroplast Genome of Salvia Sect. Drymosphace: Comparative and Phylogenetic Analysis. Diversity. 2022; 14(5):324. https://doi.org/10.3390/d14050324
Chicago/Turabian StyleSu, Ting, Yan-Fei Geng, Chun-Lei Xiang, Fei Zhao, Mei Wang, Li Gu, and Guo-Xiong Hu. 2022. "Chloroplast Genome of Salvia Sect. Drymosphace: Comparative and Phylogenetic Analysis" Diversity 14, no. 5: 324. https://doi.org/10.3390/d14050324
APA StyleSu, T., Geng, Y. -F., Xiang, C. -L., Zhao, F., Wang, M., Gu, L., & Hu, G. -X. (2022). Chloroplast Genome of Salvia Sect. Drymosphace: Comparative and Phylogenetic Analysis. Diversity, 14(5), 324. https://doi.org/10.3390/d14050324