
Survival and Genome Diversity of Vibrio parahaemolyticus Isolated from Edible Aquatic Animals



Abstract
:1. Introduction
2. Materials and Methods
2.1. V. parahaemolyticus Isolates and Culture Conditions
2.2. Genomic DNA Preparation
2.3. PCR Assay
2.4. Genome Sequencing, Assembly, and Annotation
2.5. Prediction of MGEs
2.6. Comparative Genome Analysis
3. Results
3.1. Genotypes and Phenotypes of the Six V. parahaemolyticus Isolates
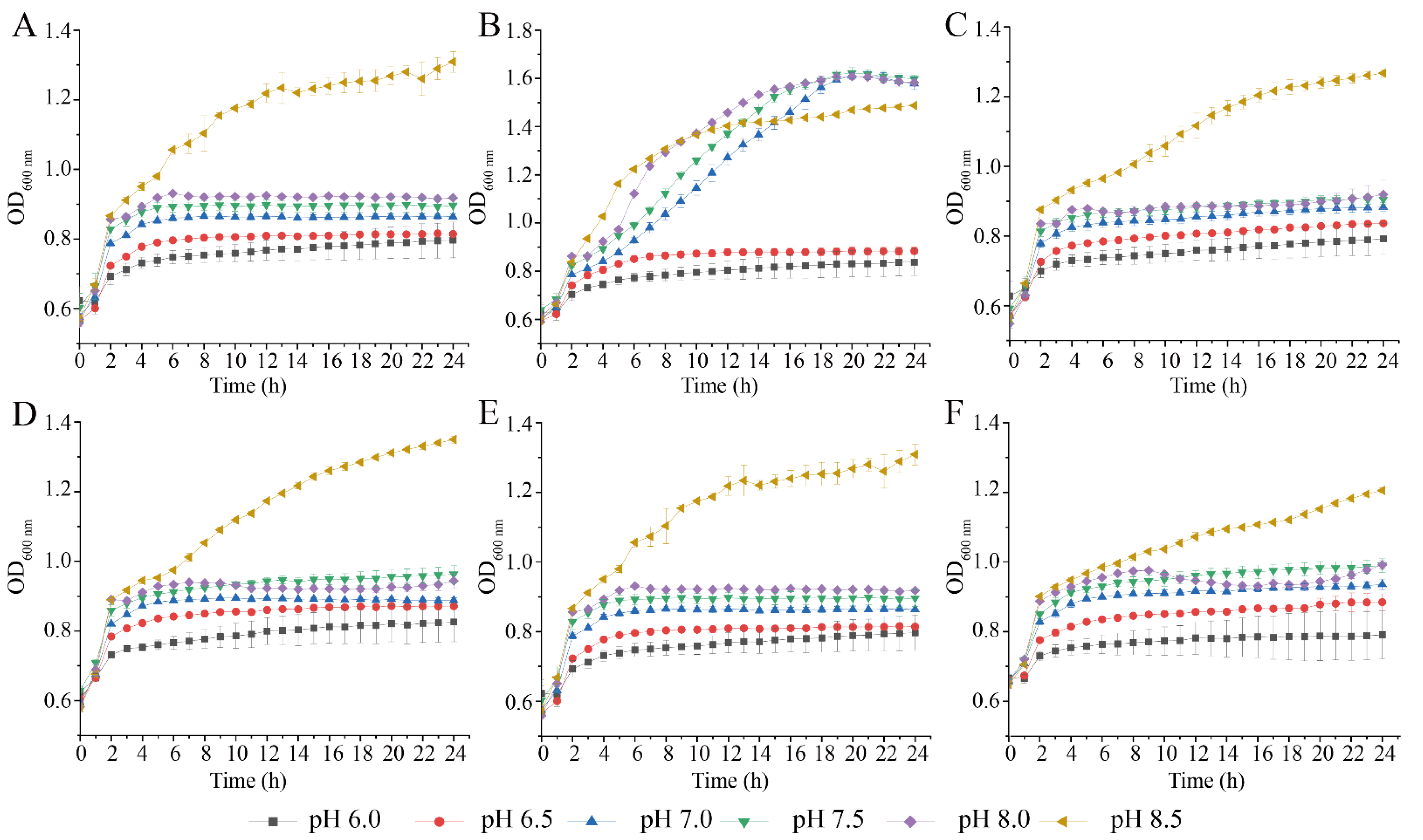
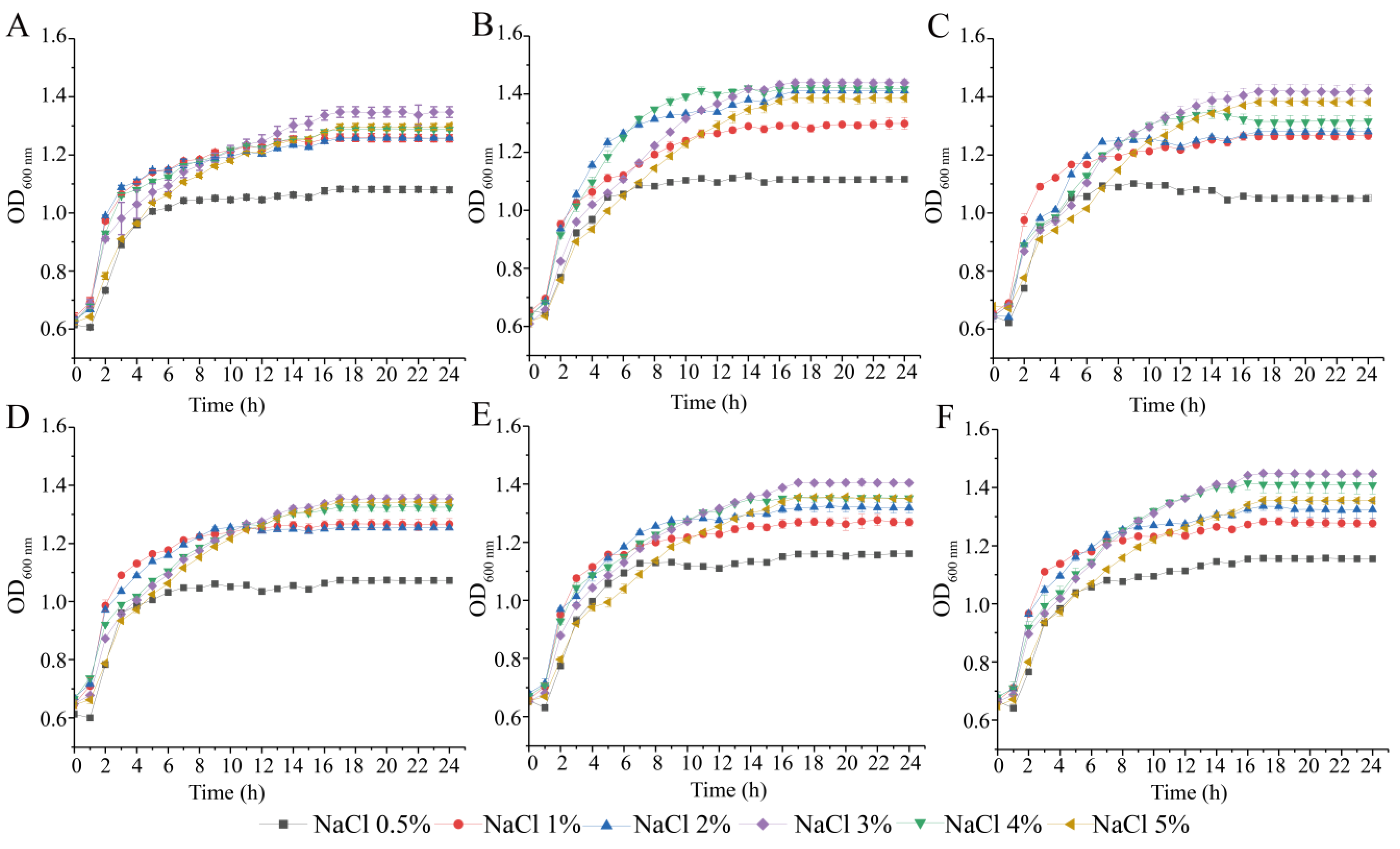
3.2. Survival of the Six V. parahaemolyticus Isolates at Different pH and NaCl Conditions
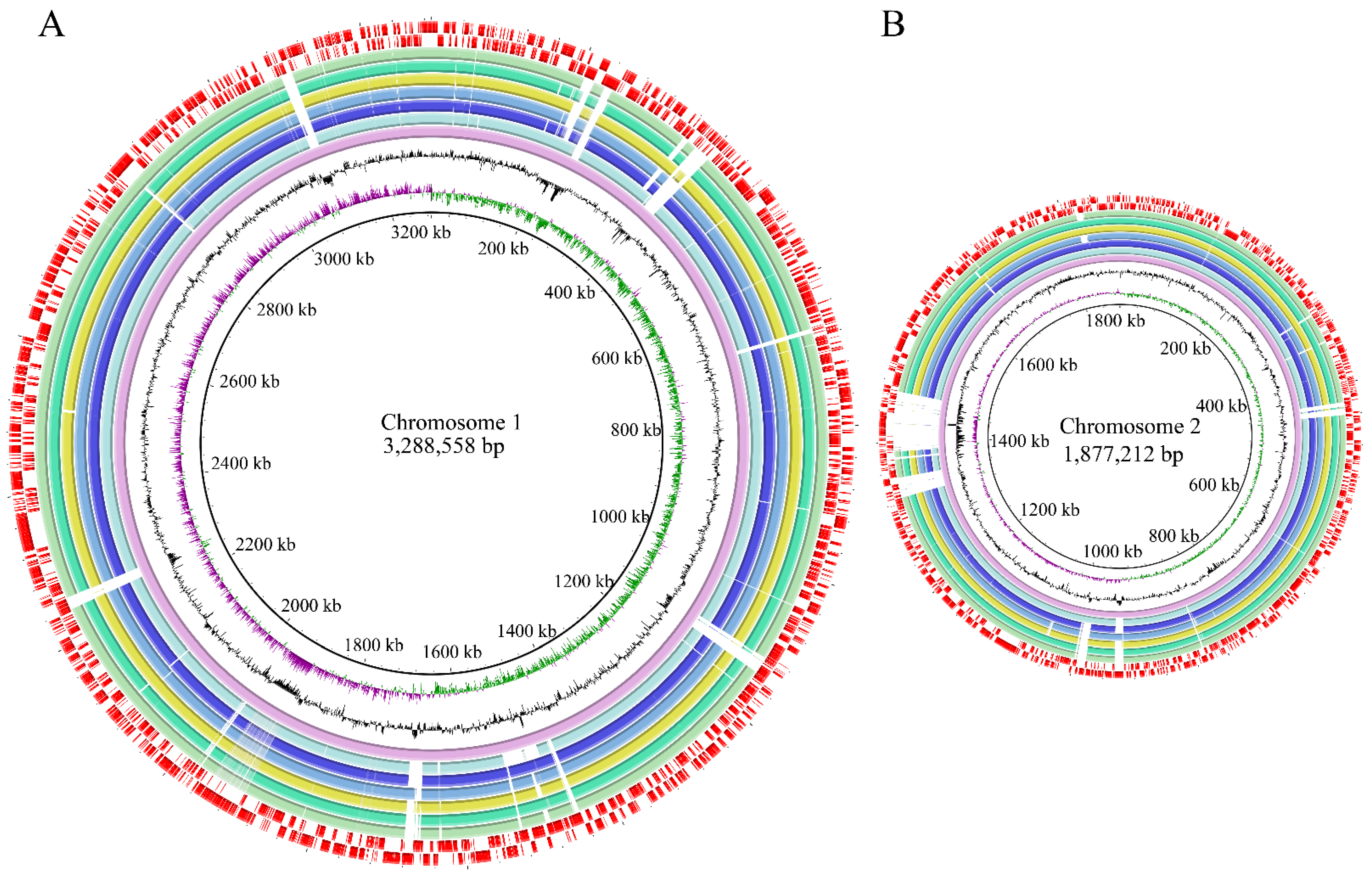
3.3. Genome Features of the Six V. parahaemolyticus Isolates Originating in Edible Aquatic Animals
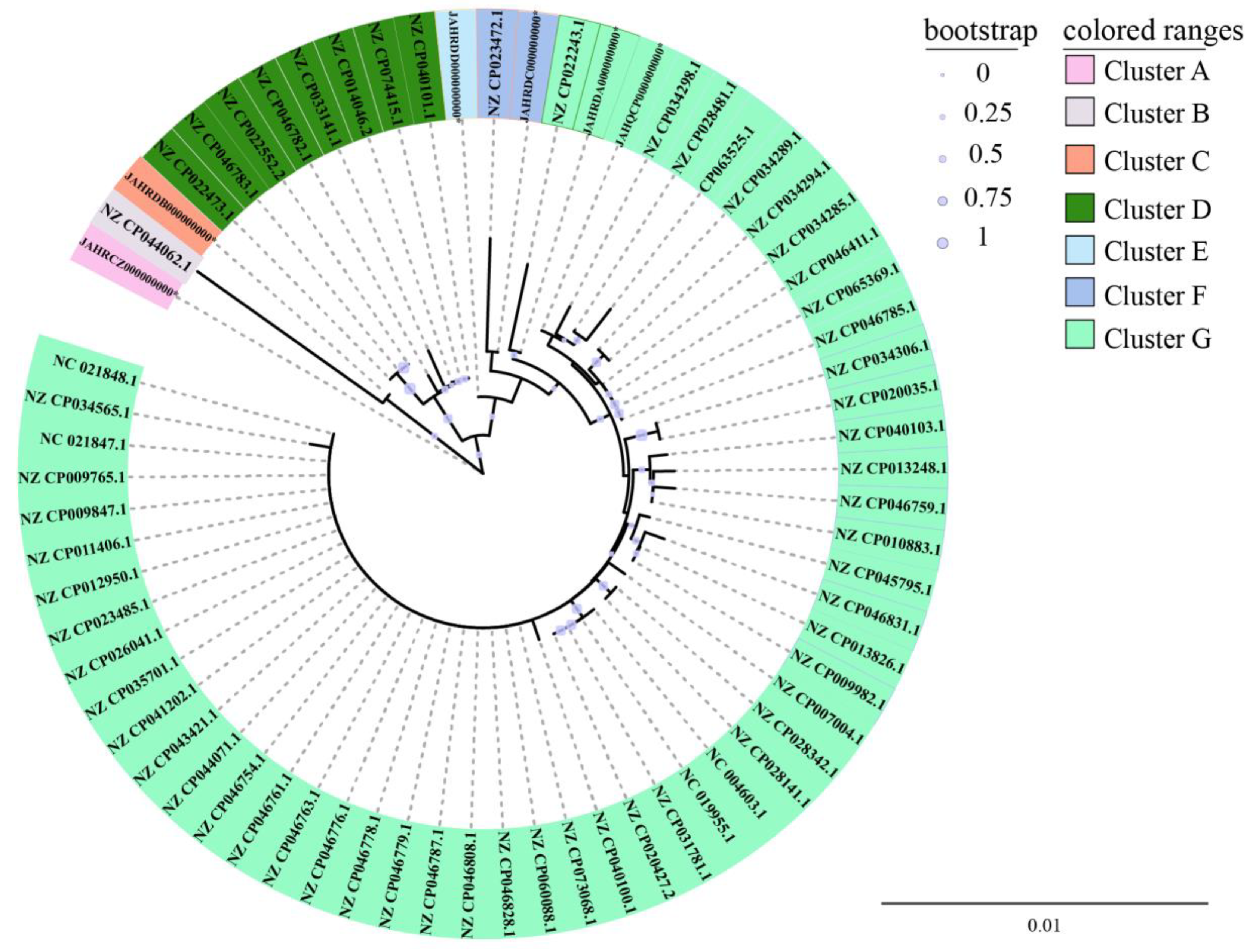
3.4. Phylogenetic Relatedness of the Six V. parahaemolyticus Isolates Originating in Edible Aquatic Animals
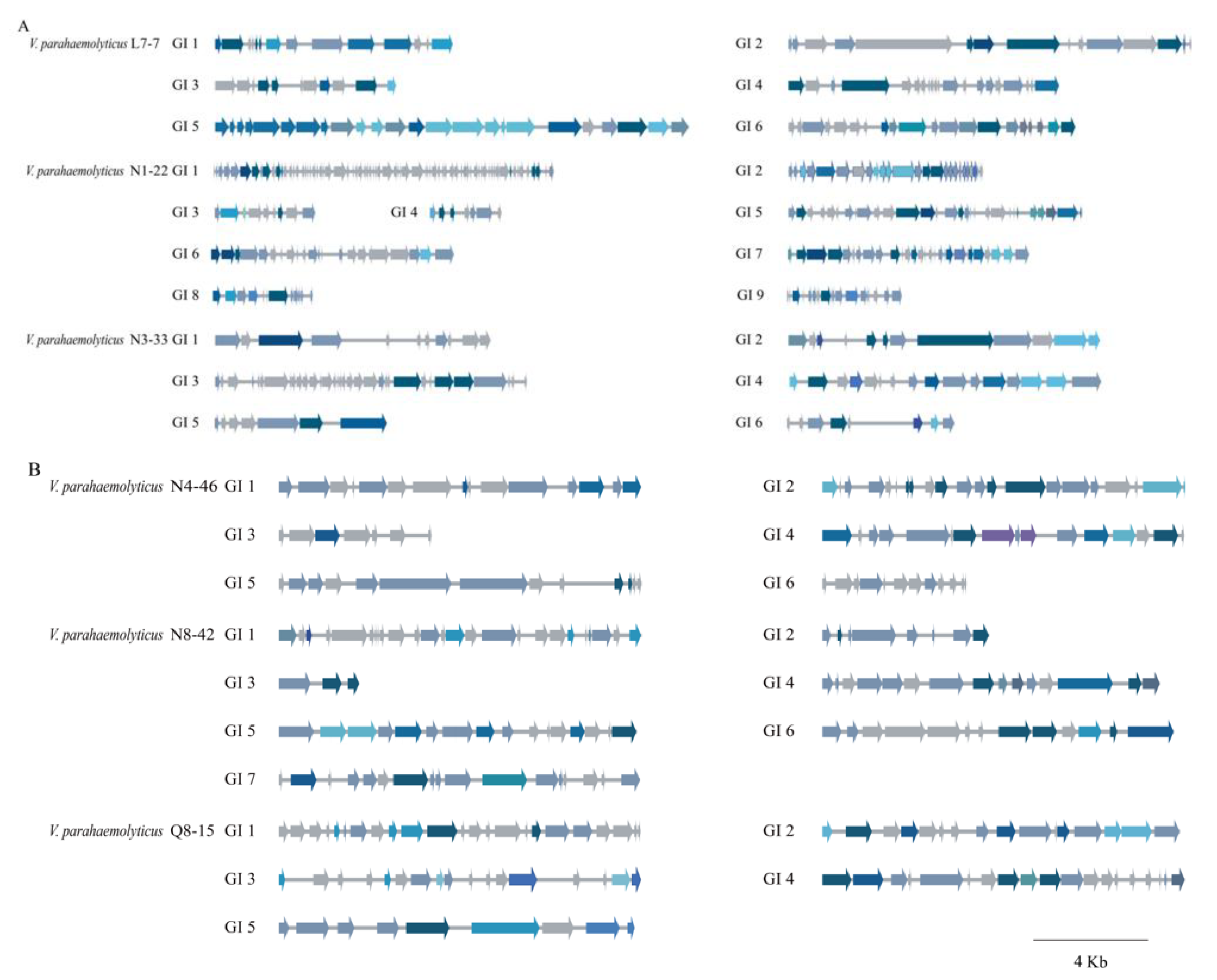
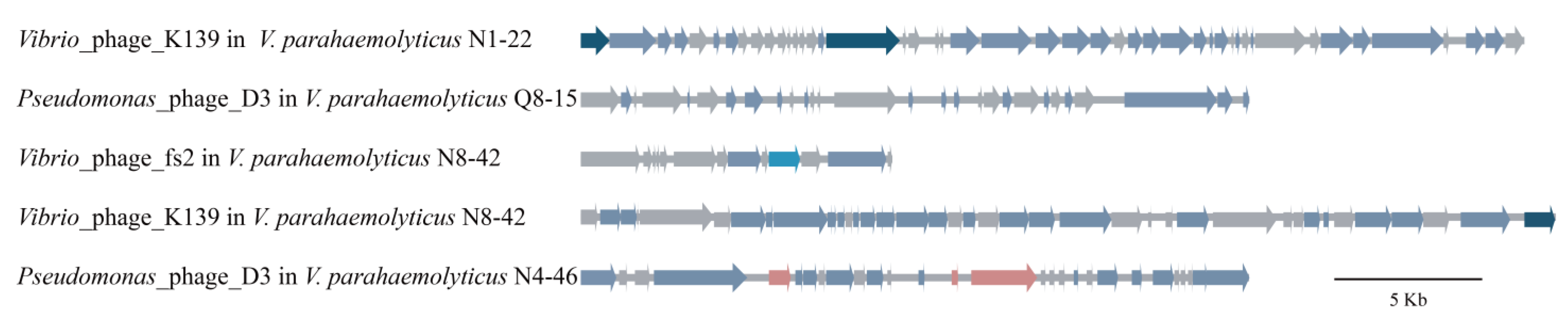
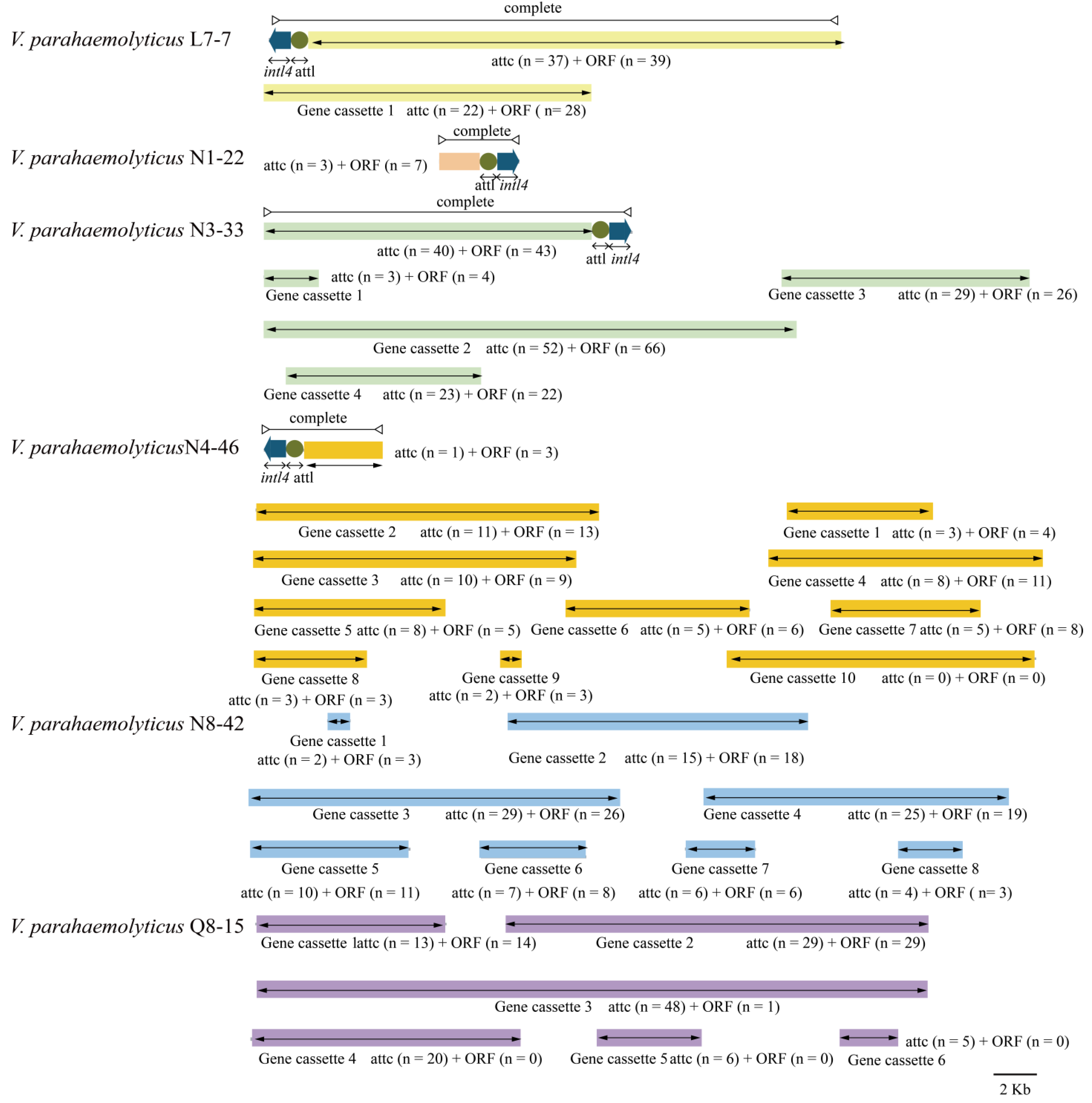
3.5. Diverse MGEs in the Six V. parahaemolyticus Genomes of Edible Aquatic Animal Origins
3.5.1. GIs
3.5.2. Prophages
3.5.3. Integrons
3.5.4. ISs
3.5.5. ICEs
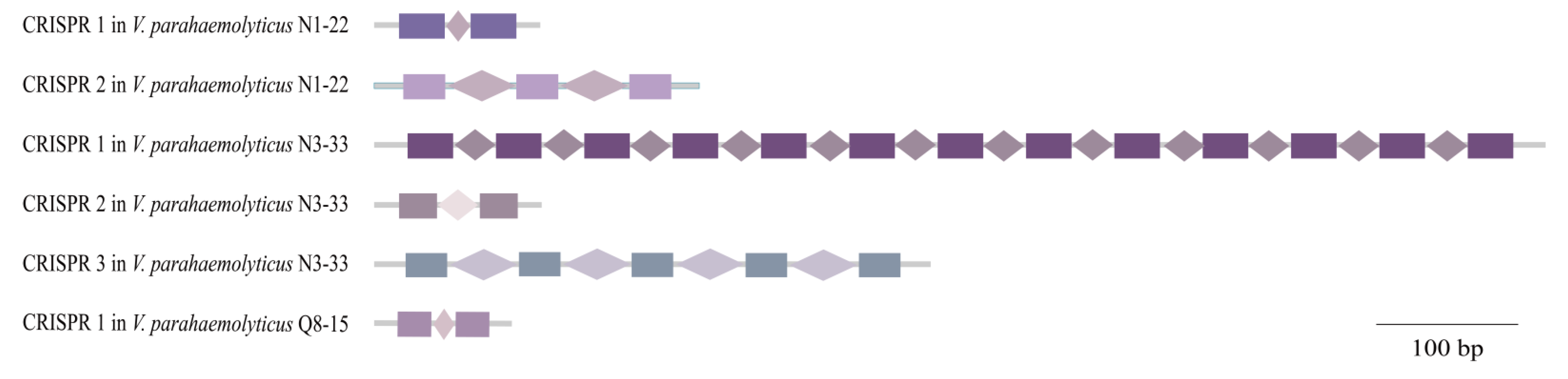
3.6. CRISPR-Cas Systems
3.7. Putative Virulence-Associated Genes in the Six V. Parahaemolyticus Genomes
3.8. Antibiotic Resistance-Associated Genes in the Six V. parahaemolyticus Genomes
3.9. Heavy Metal Tolerance-Associated Genes in the Six V. parahaemolyticus Genomes
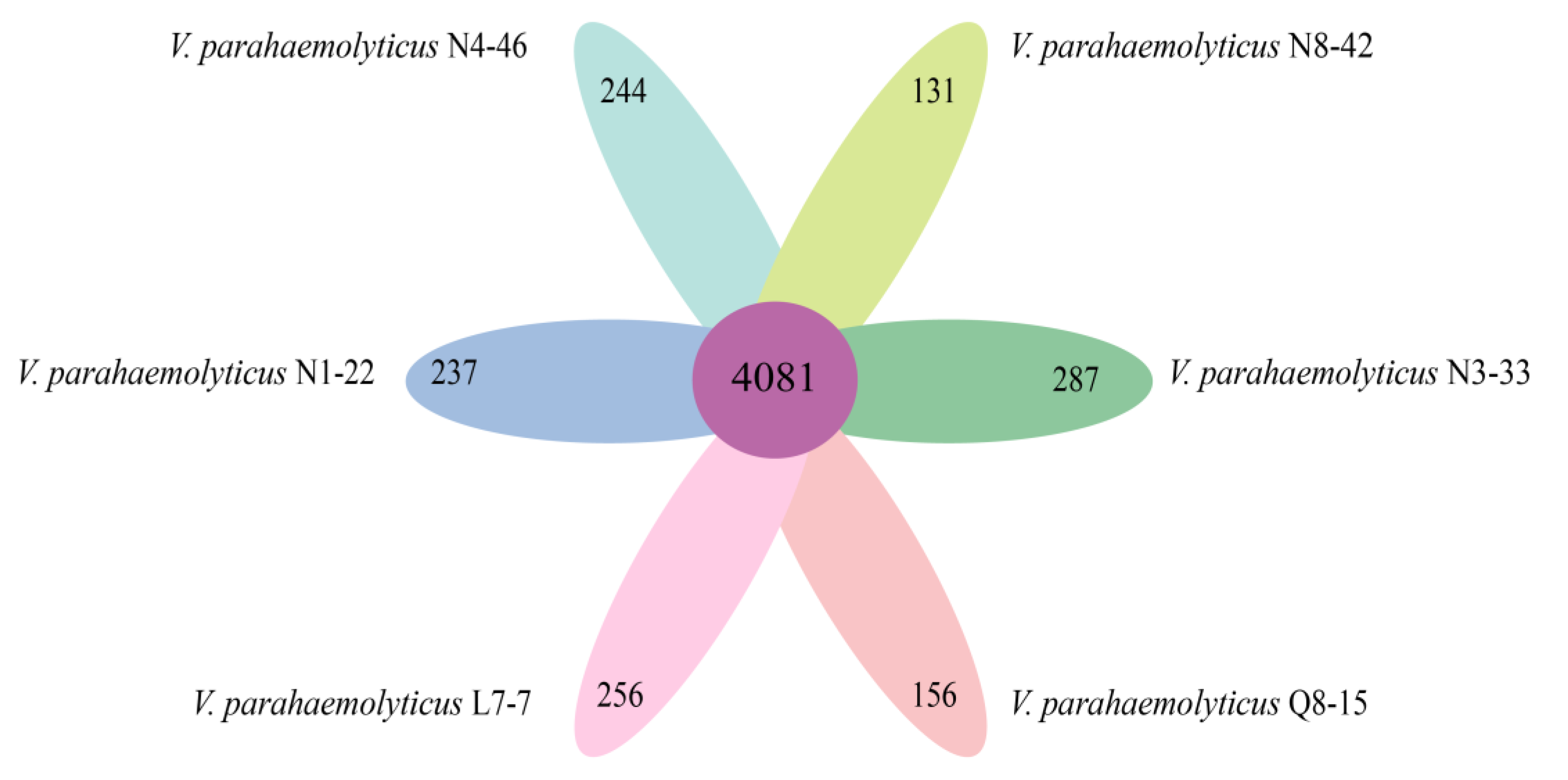
3.10. Strain-Specific Genes of the Six V. parahaemolyticus Isolates of Edible Aquatic Animal Origins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fu, S.; Wang, Q.; Zhang, Y.; Yang, Q.; Hao, J.; Liu, Y.; Pang, B. Dynamics and microevolution of Vibrio parahaemolyticus populations in shellfish farms. Msystems 2021, 6, e01161-20. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Cruz-Paredes, C.; Zhang, S.; Rousk, J. Can heavy metal pollution induce bacterial resistance to heavy metals and antibiotics in soils from an ancient land-mine. J. Hazard Mater. 2021, 411, 124962. [Google Scholar] [CrossRef] [PubMed]
- Meparambu Prabhakaran, D.; Ramamurthy, T.; Thomas, S. Genetic and virulence characterisation of Vibrio parahaemolyticus isolated from Indian coast. BMC Microbiol. 2020, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Pipes, S.; Lovell, C.R. Occurrence and significance of pathogenicity and fitness islands in environmental Vibrios. AMB Express 2018, 8, 177. [Google Scholar] [CrossRef]
- Liu, J.; Bai, L.; Li, W.; Han, H.; Fu, P.; Ma, X.; Bi, Z.; Yang, X.; Zhang, X.; Zhen, S. Trends of foodborne diseases in China: Lessons from laboratory-based surveillance since 2011. Front. Med. PRC 2018, 12, 48–57. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile genetic elements associated with antimicrobial resistance. Clin Microbiol Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [Green Version]
- Jeamsripong, S.; Khant, W.; Chuanchuen, R. Distribution of phenotypic and genotypic antimicrobial resistance and virulence genes in Vibrio parahaemolyticus isolated from cultivated oysters and estuarine water. FEMS Microbiol. Ecol. 2020, 96, fiaa081. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, L.; Yu, P.; Wang, Y.; Peng, X.; Chen, L. Comparative proteomics and secretomics revealed virulence and antibiotic resistance-associated factors in Vibrio parahaemolyticus recovered from commonly consumed aquatic products. Front. Microbiol. 2020, 11, 1453. [Google Scholar] [CrossRef]
- Coutinho, F.H.; Tschoeke, D.A.; Clementino, M.M.; Thompson, C.C.; Thompson, F.L. Genomic basis of antibiotic resistance in Vibrio parahaemolyticus strain JPA1. Mem. Inst. Oswaldo. Cruz. 2019, 114, e190053. [Google Scholar] [CrossRef]
- Li, D.; Zang, M.; Li, X.; Zhang, K.; Zhang, Z.; Wang, S. A study on the food fraud of national food safety and sample inspection of China. Food Control 2020, 116, 107306. [Google Scholar] [CrossRef]
- Ling, N.; Dailing, C.; Huiyu, F.; Qingchao, X.; Yao, L.; Xichang, W.; Yong, Z.; Lanming, C. Residual levels of antimicrobial agents and heavy metals in 41 species of commonly consumed aquatic products in Shanghai, China, and cumulative exposure risk to children and teenagers. Food Control 2021, 129, 108225. [Google Scholar] [CrossRef]
- Han, F.; Walker, R.D.; Janes, M.E.; Prinyawiwatkul, W.; Ge, B. Antimicrobial susceptibilities of Vibrio parahaemolyticus and Vibrio vulnificus isolates from Louisiana Gulf and retail raw oysters. Appl. Environ. Microbiol. 2007, 73, 7096–7098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.; Chen, L. Virulence, resistance, and genetic diversity of Vibrio parahaemolyticus recovered from commonly consumed aquatic products in Shanghai, China. Mar. Pollut. Bull. 2020, 160, 111554. [Google Scholar] [CrossRef] [PubMed]
- Fondi, M.; Karkman, A.; Tamminen, M.V.; Bosi, E.; Virta, M.; Fani, R.; Alm, E.; McInerney, J.O. “Every gene is everywhere but the environment selects”: Global geolocalization of gene sharing in environmental samples through network analysis. Genome Biol. Evol. 2016, 8, 1388–1400. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gu, J.; Wang, X.; Ma, J.; Hu, T.; Peng, H.; Bao, J.; Zhang, R. Effects of nano-zerovalent iron on antibiotic resistance genes and mobile genetic elements during swine manure composting. Environ. Pollut. 2020, 258, 113654. [Google Scholar] [CrossRef]
- Dionisio, F.; Zilhão, R.; Gama, J.A. Interactions between plasmids and other mobile genetic elements affect their transmission and persistence. Plasmid 2019, 102, 29–36. [Google Scholar] [CrossRef]
- Thomas, C.M.; Nielsen, K.M. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 2005, 3, 711–721. [Google Scholar] [CrossRef]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef]
- Canchaya, C.; Fournous, G.; Chibani-Chennoufi, S.; Dillmann, M.L.; Brüssow, H. Phage as agents of lateral gene transfer. Curr. Opin. Microbiol. 2003, 6, 417–424. [Google Scholar] [CrossRef]
- Metzker, M.L. Emerging technologies in DNA sequencing. Genome Res. 2005, 15, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- García, K.; Yáñez, C.; Plaza, N.; Peña, F.; Sepúlveda, P.; Pérez-Reytor, D.; Espejo, R.T. Gene expression of Vibrio parahaemolyticus growing in laboratory isolation conditions compared to those common in its natural ocean environment. BMC Microbiol. 2017, 17, 118. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.W.; Rukayadi, Y.; Hasan, H.; Thung, T.Y.; Lee, E.; Rollon, W.D.; Hara, H.; Kayali, A.Y.; Nishibuchi, M.; Radu, S. Prevalence and antibiotic resistance patterns of Vibrio parahaemolyticus isolated from different types of seafood in Selangor, Malaysia. Saudi J. Biol. Sci. 2020, 27, 1602–1608. [Google Scholar] [CrossRef]
- Sun, X.; Liu, T.; Peng, X.; Chen, L. Insights into Vibrio parahaemolyticus CHN25 response to artificial gastric fluid stress by transcriptomic analysis. Int. J. Mol. Sci. 2014, 15, 22539–22562. [Google Scholar] [CrossRef]
- Zhu, C.; Sun, B.; Liu, T.; Zheng, H.; Gu, W.; He, W.; Sun, F.; Wang, Y.; Yang, M.; Bei, W.; et al. Genomic and transcriptomic analyses reveal distinct biological functions for cold shock proteins (VpaCspA and VpaCspD) in Vibrio parahaemolyticus CHN25 during low-temperature survival. BMC Genom. 2017, 18, 436. [Google Scholar] [CrossRef] [Green Version]
- Wenting, Y.; Lianzhi, Y.; Zehuai, S.; Lu, X.; Lanming, C. Identification of salt tolerance-related genes of Lactobacillus plantarum D31 and T9 strains by genomic analysis. Ann. Microbiol. 2020, 70, 10. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, Y.; Yu, P.; Ren, S.; Zhu, Z.; Jin, Y.; Yan, J.; Peng, X.; Chen, L. Prophage-related gene VpaChn25_0724 contributes to cell membrane integrity and growth of Vibrio parahaemolyticus CHN25. Front. Cell. Infect. Microbiol. 2020, 10, 595709. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. EggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008, 36, D250–D254. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Cury, J.; Jové, T.; Touchon, M.; Néron, B.; Rocha, E.P. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic Acids Res. 2016, 44, 4539–4550. [Google Scholar] [CrossRef] [Green Version]
- Siguier, P.; Gourbeyre, E.; Chandler, M. Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev. 2014, 38, 865–891. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Yu, P.; Zheng, H.; Gu, W.; He, W.; Tang, Y.; Wang, Y.; Dong, Y.; Peng, X.; She, Q.; et al. Comparative genomics for non-O1/O139 Vibrio cholerae isolates recovered from the Yangtze River Estuary versus V. cholerae representative isolates from serogroup O1. Mol. Genet. Genom. 2019, 294, 417–430. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Kong, R.; Xu, X.; Liu, X.; He, P.; Zhang, M.Q.; Dai, Q. 2SigFinder: The combined use of small-scale and large-scale statistical testing for genomic island detection from a single genome. BMC Bioinform. 2020, 21, 159. [Google Scholar] [CrossRef]
- Onesime, M.; Yang, Z.; Dai, Q. Genomic island prediction via chi-square test and random forest algorithm. Comput. Math. Methods Med. 2021, 2021, 9969751. [Google Scholar] [CrossRef]
- Liu, P.; Huang, D.; Hu, X.; Tang, Y.; Ma, X.; Yan, R.; Han, Q.; Guo, J.; Zhang, Y.; Sun, Q.; et al. Targeting inhibition of smpB by peptide aptamer attenuates the virulence to protect zebrafish against Aeromonas veronii infection. Front. Microbiol. 2017, 8, 1766. [Google Scholar] [CrossRef]
- Lee, H.A.; Kim, J.Y.; Kim, J.; Nam, B.; Kim, O. Anti-helicobacter pylori activity of acomplex mixture of Lactobacillus paracasei HP7 including the extract of perilla frutescens var, acuta and glycyrrhiza glabra. Lab. Anim. Res. 2020, 36, 40. [Google Scholar] [CrossRef]
- Heidelberg, J.F.; Eisen, J.A.; Nelson, W.C.; Clayton, R.A.; Gwinn, M.L.; Dodson, R.J.; Haft, D.H.; Hickey, E.K.; Peterson, J.D.; Umayam, L.; et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 2000, 406, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Garin-Fernandez, A.; Wichels, A. Looking for the hidden: Characterization of lysogenic phages in potential pathogenic Vibrio species from the North Sea. Mar. Genom. 2020, 51, 100725. [Google Scholar] [CrossRef]
- Llanos Salinas, S.P.; Castillo Sánchez, L.O.; Castañeda Miranda, G.; Rodríguez Reyes, E.A.; Ordoñez López, L.; Mena Bañuelos, R.; Alcaraz Sosa, L.E.; Núñez Carrera, M.G.; José Manuel, R.O.; Carmona Gasca, C.A.; et al. GspD, the type II secretion system secretin of Leptospira, protects hamsters against lethal infection with a virulent L. interrogans isolate. Vaccines 2020, 8, 759. [Google Scholar] [CrossRef]
- Bisht, S.C.; Joshi, G.K.; Mishra, P.K. CspA encodes a major cold shock protein in Himalayan psychrotolerant Pseudomonas strains. Interdiscip. Sci. 2014, 6, 140–148. [Google Scholar] [CrossRef]
- An, X.L.; Chen, Q.L.; Zhu, D.; Zhu, Y.G.; Gillings, M.R.; Su, J.Q. Impact of wastewater treatment on the prevalence of integrons and the genetic diversity of integron gene cassettes. Appl. Environ. Microb. 2018, 84, e02766-17. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, P.; Rajabnia, M.; Maali, A.; Ferdosi-Shahandashti, E. Integron and its role in antimicrobial resistance: A literature review on some bacterial pathogens. Iran. J. Basic Med. Sci. 2021, 24, 136–142. [Google Scholar] [CrossRef]
- Vaisvila, R.; Morgan, R.D.; Posfai, J.; Raleigh, E.A. Discovery and distribution of super-integrons among pseudomonads. Mol. Microbiol. 2001, 42, 587–601. [Google Scholar] [CrossRef]
- Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Type II toxin-antitoxin systems: Evolution and revolutions. J. Bacteriol. 2020, 202, e00763-19. [Google Scholar] [CrossRef] [Green Version]
- Lux, T.M.; Lee, R.; Love, J. Genome-wide phylogenetic analysis of the pathogenic potential of Vibrio furnissii. Front. Microbiol. 2014, 5, 435. [Google Scholar] [CrossRef] [Green Version]
- Mahillon, J.; Chandler, M. Insertion sequences. Microbiol. Mol. Biol. Rev. 1998, 62, 725–774. [Google Scholar] [CrossRef] [Green Version]
- McDonald, N.D.; Regmi, A.; Morreale, D.P.; Borowski, J.D.; Boyd, E.F. CRISPR-Cas systems are present predominantly on mobile genetic elements in Vibrio species. BMC Genom. 2019, 20, 105. [Google Scholar] [CrossRef] [Green Version]
- Hensel, M.; Shea, J.E.; Raupach, B.; Monack, D.; Falkow, S.; Gleeson, C.; Kubo, T.; Holden, D.W. Functional analysis of ssaJ and the ssaK/U operon, 13 genes encoding components of the type III secretion apparatus of Salmonella pathogenicity island 2. Mol. Microbiol. 1997, 24, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, D.F.; Zhou, X.; Xu, L.; Zhang, L.; Shi, X. Comprehensive analysis reveals two distinct evolution patterns of Salmonella Flagellin gene clusters. Front. Microbiol. 2017, 8, 2604. [Google Scholar] [CrossRef]
- Martin, R.M.; Bachman, M.A. Colonization, infection, and the accessory genome of Klebsiella pneumoniae. Front. Cell. Infect. Microbiol. 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Domínguez-Bernal, G.; Pucciarelli, M.G.; Ramos-Morales, F.; García-Quintanilla, M.; Cano, D.A.; Casadesús, J.; García-del Portillo, F. Repression of the RcsC-YojN-RcsB phosphorelay by the IgaA protein is a requisite for Salmonella virulence. Mol. Microbiol. 2004, 53, 1437–1449. [Google Scholar] [CrossRef]
- Pillay, S.; Zishiri, O.T.; Adeleke, M.A. Prevalence of virulence genes in Enterococcus species isolated from companion animals and livestock. Onderstepoort J. Vet. Res. 2018, 85, e1–e8. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Nybom, P.; Magnusson, K.E. Distinct effects of Vibrio cholerae haemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell Microbiol. 2000, 2, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Kobylka, J.; Kuth, M.S.; Müller, R.T.; Geertsma, E.R.; Pos, K.M. AcrB: A mean, keen, drug efflux machine. Ann. N. Y. Acad. Sci. 2020, 1459, 38–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Szewczyk, P.; Karyakin, A.; Evin, M.; Hong, W.X.; Zhang, Q.; Chang, G. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 2010, 467, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Lin, H.C.; Huang, Y.W.; Chung, T.C.; Yang, T.C. Inactivation of mrcA gene derepresses the basal-level expression of L1 and L2 β-lactamases in Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 2011, 66, 2033–2037. [Google Scholar] [CrossRef] [Green Version]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Garg, S.; Gupta, L.; Gupta, K.; Diagne, C.T.; Missé, D.; Pompon, J.; Kumar, S.; Saxena, V. Delineating the role of aedes aegypti ABC transporter gene family during mosquito development and arboviral infection via transcriptome analyses. Pathogens 2021, 10, 1127. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Lee, S.-Y. Characteristics of viable-but-nonculturable Vibrio parahaemolyticus induced by nutrient-deficiency at cold temperature. Crit. Rev. Food Sci. 2020, 60, 1302–1320. [Google Scholar] [CrossRef]
- Matsuda, S.; Hiyoshi, H.; Tandhavanant, S.; Kodama, T. Advances on Vibrio parahaemolyticus research in the postgenomic era. Microbiol. Immunol. 2020, 64, 167–181. [Google Scholar] [CrossRef]
- Pazhani, G.P.; Chowdhury, G.; Ramamurthy, T. Adaptations of Vibrio parahaemolyticus to stress during environmental survival, host colonization, and infection. Front. Microbiol. 2021, 12, 737299. [Google Scholar] [CrossRef]
- Magaziner, S.J.; Zeng, Z.; Chen, B.; Salmond, G.P.C. The prophages of citrobacter rodentium represent a conserved family of horizontally acquired mobile genetic elements associated with enteric evolution towards pathogenicity. J. Bacteriol. 2019, 201, e00638-18. [Google Scholar] [CrossRef] [Green Version]
- Ridder, M.J.; Daly, S.M.; Hall, P.R.; Bose, J.L. Quantitative hemolysis assays. Methods Mol. Biol. 2021, 2341, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Buongermino Pereira, M.; Österlund, T.; Eriksson, K.M.; Backhaus, T.; Axelson-Fisk, M.; Kristiansson, E. A comprehensive survey of integron-associated genes present in metagenomes. BMC Genom. 2020, 21, 495. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bansal, K.; Patil, P.P.; Kaur, A.; Kaur, S.; Jaswal, V.; Gautam, V.; Patil, P.B. Genomic insights into evolution of extensive drug resistance in Stenotrophomonas maltophilia complex. Genomics 2020, 112, 4171–4178. [Google Scholar] [CrossRef] [PubMed]
- Rowe-Magnus, D.A.; Guérout, A.M.; Mazel, D. Super-integrons. Res. Microbiol. 1999, 150, 641–651. [Google Scholar] [CrossRef]
- Gonçalves, O.S.; Campos, K.F.; de Assis, J.C.S.; Fernandes, A.S.; Souza, T.S.; do Carmo Rodrigues, L.G.; Queiroz, M.V.; Santana, M.F. Transposable elements contribute to the genome plasticity of Ralstonia solanacearum species complex. Microb. Genom. 2020, 6, e000374. [Google Scholar] [CrossRef]
- Mackow, N.A.; Shen, J.; Adnan, M.; Khan, A.S.; Fries, B.C.; Diago-Navarro, E. CRISPR-Cas influences the acquisition of antibiotic resistance in Klebsiella pneumoniae. PLoS ONE 2019, 14, e0225131. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Makarova, K.S. Origins and evolution of CRISPR-Cas systems. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180087. [Google Scholar] [CrossRef]
- Castillo, D.; Pérez-Reytor, D.; Plaza, N.; Ramírez-Araya, S.; Blondel, C.J.; Corsini, G.; Bastías, R.; Loyola, D.E.; Jaña, V.; Pavez, L.; et al. Exploring the genomic traits of non-toxigenic Vibrio parahaemolyticus strains isolated in southern chile. Front. Microbiol. 2018, 9, 161. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, E.; Rehman, A.; Ali, M.A. Validation of serum C-reactive protein for the diagnosis and monitoring of antibiotic therapy in neonatal sepsis. Pak. J. Med. Sci. 2017, 33, 1434–1437. [Google Scholar] [CrossRef]
- El-Razik, K.A.A.; Arafa, A.A.; Hedia, R.H.; Ibrahim, E.S. Tetracycline resistance phenotypes and genotypes of coagulase-negative staphylococcal isolates from bubaline mastitis in Egypt. Vet. World. 2017, 10, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Shin, C.; Shin, Y.; Kim, P.H.; Park, J.I.; Kim, M.; Park, B.; So, J.S. Heavy metal and antibiotic co-resistance in Vibrio parahaemolyticus isolated from shellfish. Mar. Pollut. Bull. 2020, 156, 111246. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Feature | V. parahaemolyticus Isolate | |||||
|---|---|---|---|---|---|---|
| L7-7 | N1-22 | N3-33 | N4-46 | N8-42 | Q8-15 | |
| Genome size (bp) | 4,937,042 | 5,067,778 | 5,018,989 | 5,049,514 | 5,010,476 | 4,993,599 |
| G + C (%) | 45.36 | 45.20 | 45.32 | 45.29 | 45.39 | 45.34 |
| DNA Scaffold | 54 | 58 | 60 | 77 | 61 | 36 |
| Total predicted gene | 4749 | 4919 | 4815 | 4878 | 4832 | 4756 |
| Protein-coding gene | 4624 | 4791 | 4707 | 4768 | 4706 | 4622 |
| RNA gene | 125 | 128 | 108 | 110 | 126 | 134 |
| Genes assigned to COG | 3862 | 3892 | 3852 | 3886 | 3875 | 3854 |
| Genes with unknown function | 1064 | 1094 | 1086 | 1107 | 1102 | 1093 |
| GI | 6 | 9 | 6 | 6 | 7 | 5 |
| Prophage gene cluster | 0 | 1 | 0 | 1 | 2 | 1 |
| CRISPR-Cas | 0 | 2 | 3 | 0 | 0 | 1 |
| Integron | 2 | 1 | 5 | 11 | 8 | 6 |
| IS | 3 | 0 | 1 | 0 | 1 | 1 |
| 16S rDNA sequence accession no. | MW386441 | MW386442 | MW386445 | MW386446 | MW386450 | MW386453 |
| Antimicrobial Agent | Gene | V. parahaemolyticus Isolates |
|---|---|---|
| Cephalosporin | acrB | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| Phenicol | catB | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| SoxR | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 | |
| Fluoroquinolone | hns | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| acrB | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 | |
| qnr | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 | |
| crp | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 | |
| Tetracycline | tet(35) | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| tet(34) | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 | |
| hns | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 | |
| acrB | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 | |
| SoxR | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 | |
| Beta-lactam | blaCARB-18 | N3-33 |
| blaCARB-19 | L7-7, N1-22, N8-42, Q8-15 | |
| blaCARB-21 | L7-7, N1-22, N3-33, N8-42, Q8-15 | |
| blaCARB-23 | N4-46 | |
| blaCARB-29 | N3-33, N4-46,Q8-15 | |
| blaCARB-35 | N1-22 | |
| blaCARB-33 | N3-33 | |
| blaCARB-41 | N3-33 | |
| blaCARB-44 | L7-7, N8-42 | |
| blaCARB-45 | N1-22 | |
| blaCARB-48 | N1-22 | |
| Diaminopyrimidine | folA | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| Macrolide | macB | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| Nitroimidazole | msbA | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| Heavy metal | cusA | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| Heavy metal | cusR | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| Heavy metal | cusS | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| Heavy metal | zntA | L7-7, N1-22, N3-33, N4-46, N8-42, Q8-15 |
| Heavy metal | copA | L7-7, Q8-15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, D.; Peng, X.; Xie, L.; Chen, L. Survival and Genome Diversity of Vibrio parahaemolyticus Isolated from Edible Aquatic Animals. Diversity 2022, 14, 350. https://doi.org/10.3390/d14050350
Xu D, Peng X, Xie L, Chen L. Survival and Genome Diversity of Vibrio parahaemolyticus Isolated from Edible Aquatic Animals. Diversity. 2022; 14(5):350. https://doi.org/10.3390/d14050350
Chicago/Turabian StyleXu, Dingxiang, Xu Peng, Lu Xie, and Lanming Chen. 2022. "Survival and Genome Diversity of Vibrio parahaemolyticus Isolated from Edible Aquatic Animals" Diversity 14, no. 5: 350. https://doi.org/10.3390/d14050350
APA StyleXu, D., Peng, X., Xie, L., & Chen, L. (2022). Survival and Genome Diversity of Vibrio parahaemolyticus Isolated from Edible Aquatic Animals. Diversity, 14(5), 350. https://doi.org/10.3390/d14050350