
Advances in Nematode Identification: A Journey from Fundamentals to Evolutionary Aspects

 ,
,  ,
,  ,
,
Abstract
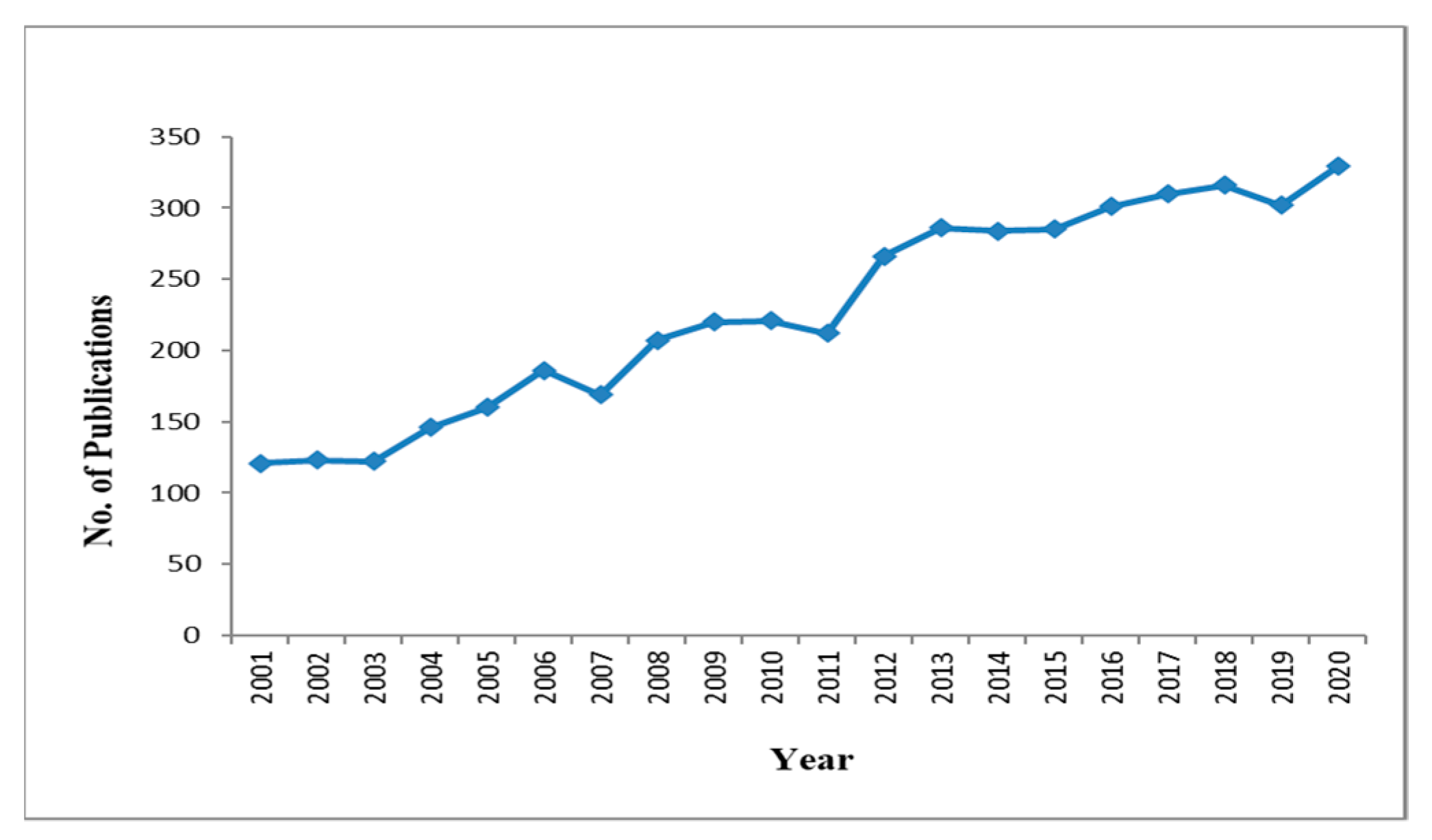
:1. Introduction
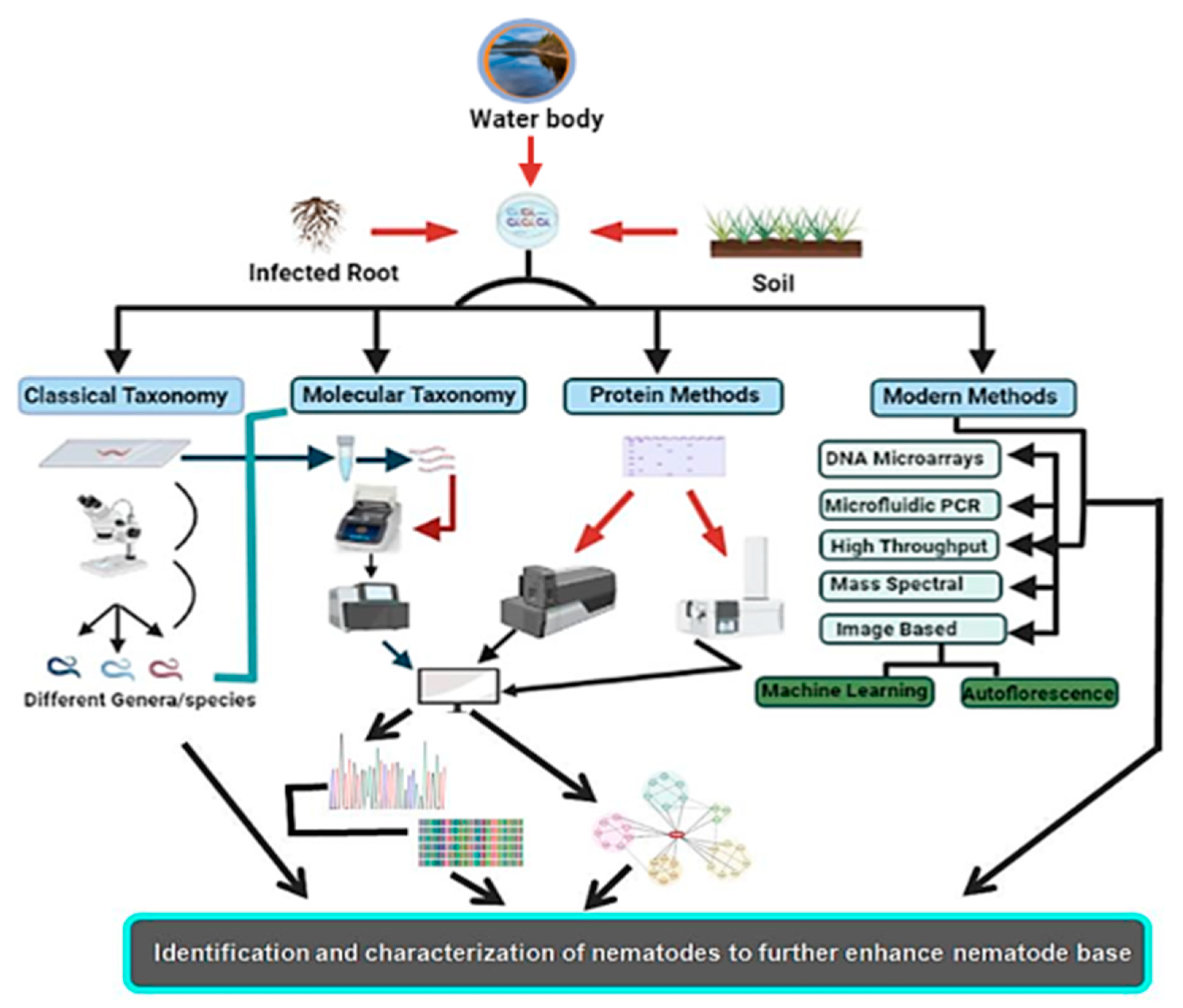
2. Conventional or Morphometric Method of Identification
Morphological Methods of Identification
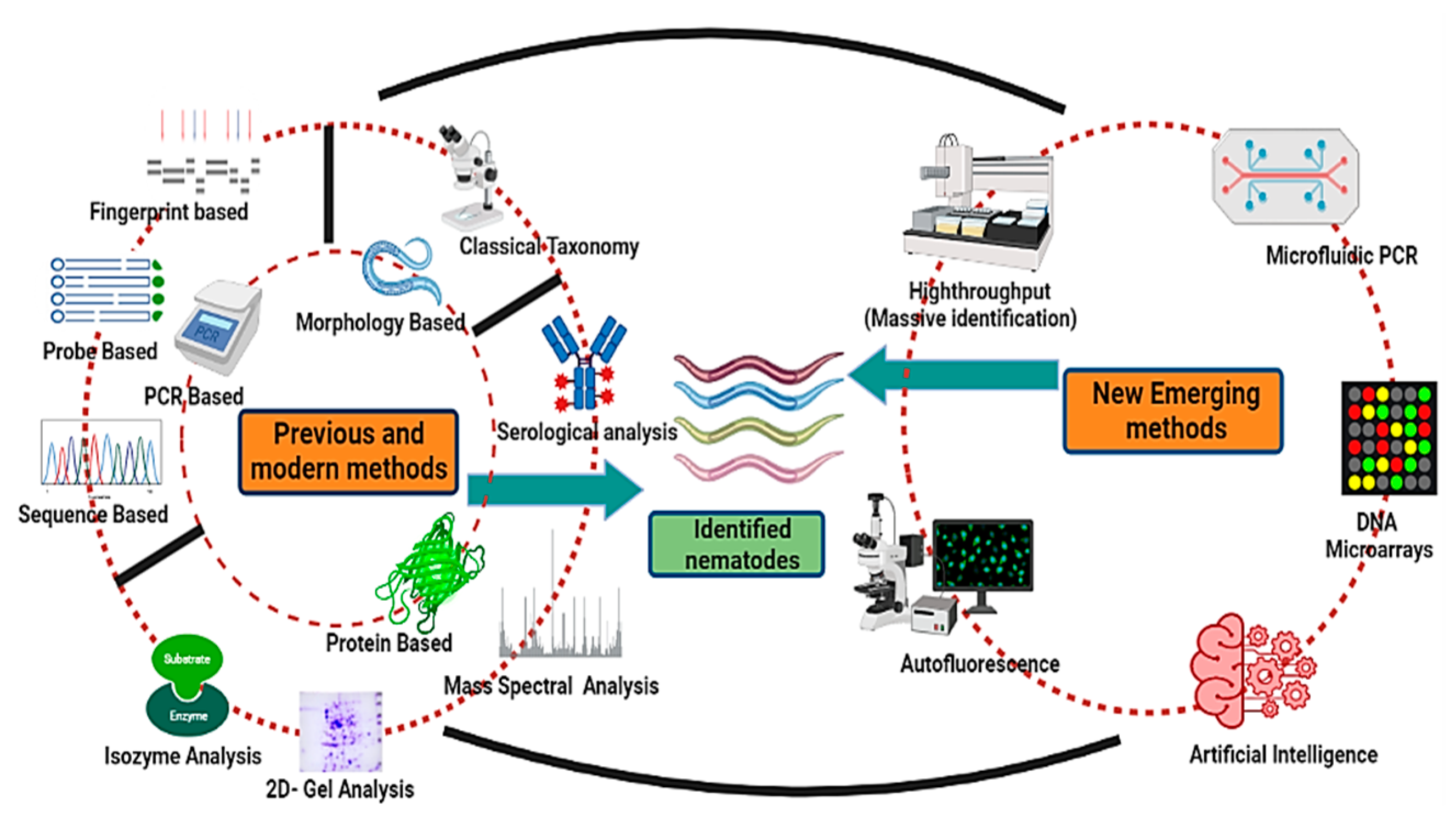
3. Molecular Identification Methods
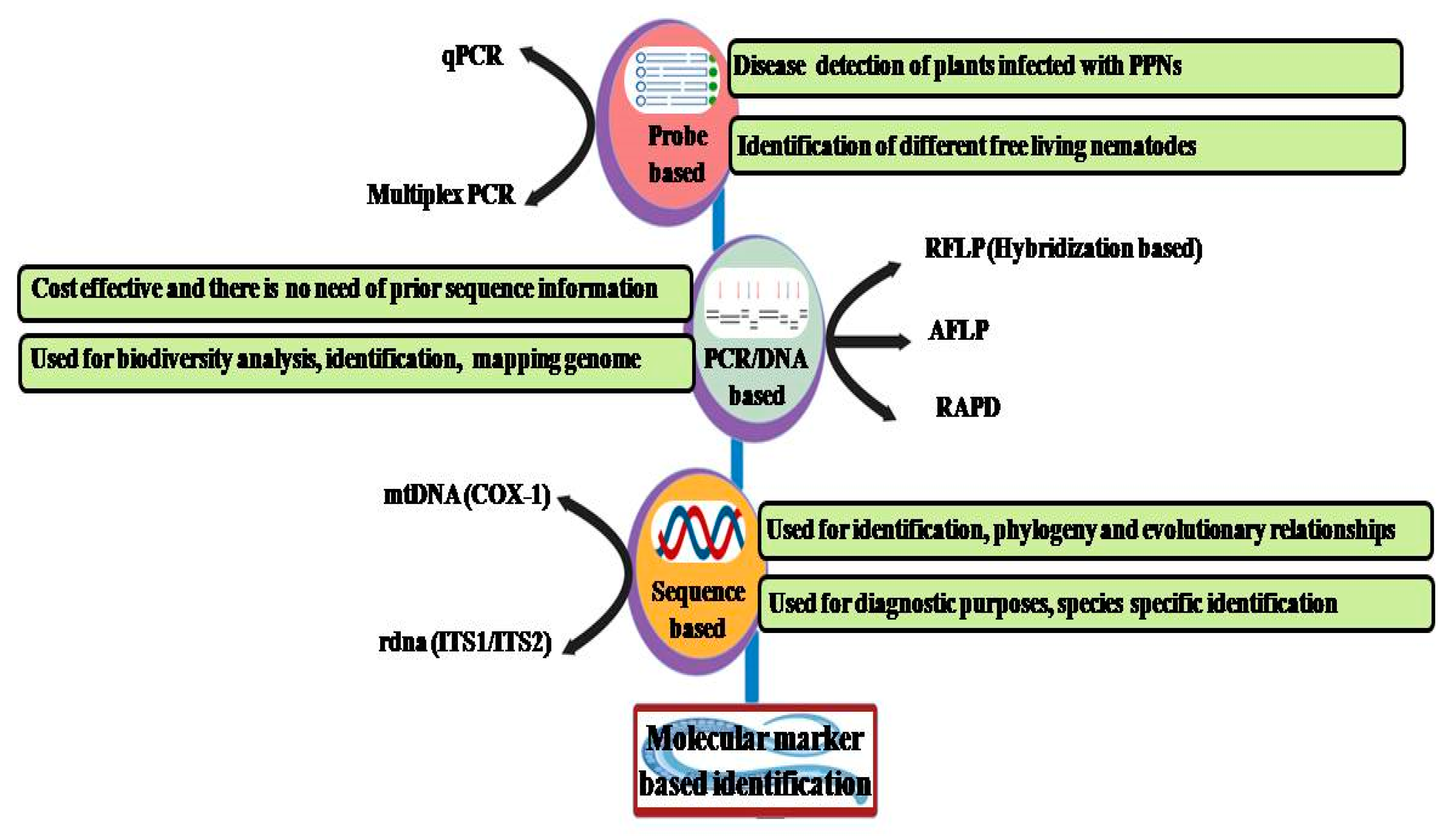
3.1. PCR-Based Methods
3.2. Fingerprint-Based Methods
3.2.1. RFLP (Restriction Fragment Length Polymorphism)
3.2.2. Random Amplified Polymorphic DNA (RAPD)
3.2.3. Amplified Fragment Length Polymorphism (AFLP)
3.3. Probe-Based Detection Methods
3.4. Sequence-Based Detection Method
4. Metabarcoding

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Target | Resolution | Aim | Cost | Pros | Cons | Reference | |
|---|---|---|---|---|---|---|---|---|
| Morphological-Based | ||||||||
| Classical Morphometrics | Whole organism | Medium | Re-description of recognized species, and a description of new species | Low | Timeless and simple setup required; cost effective | No distinguishable morphological traits such as in larvae Paucity of trained taxonomists | [4,14,15,20] | |
| Molecular/DNA-Based | ||||||||
| Fingerprint/Hybridization-Based | RFLP | DNA | Medium | Known species identification | Medium | Identification of related species | Time-consuming | [72] |
| RAPD | DNA | Medium | Species identification. Phylogeny between species | Medium | Reproducibility. Prior information on the sequence is not required. A small amount of DNA is needed | Sensitivity Time-consuming | [14] | |
| AFLP | DNA | Medium | Species identification. Gene expression analysis and study | Medium | Prior information on the sequence is not required. A small amount of DNA is needed | Time-consuming. Individual identification not possible | [39,73] | |
| Probe-Based | Multiplex-PCR | DNA | Low | Cryptic Species identification | Medium | Simultaneous study of several target genes. Time effective | Same primer but different target genes. Low-throughput identification | [42,43] |
| Real time PCR (qPCR) | DNA | Medium | Cryptic Species identification | High | Polymorphism detection | Cost factor Low throughput | [45,46,48] | |
| Sequence-Based | ITS | rDNA | High | Identification of cryptic or sibling species | High | Reference material and data | Automated processes limitation. Individual Identification | [29,74] |
| COX | DNA | High | Identification of unknown species | High | Reference material and data | Automated processes limitation. Individual identification, standardization | [15] | |
| Protein/Biochemical-Based | ||||||||
| Isozyme Analysis | Intracellular enzymes | Medium | Identification of known species and description of new species | Medium | Better performance; cost- and time-effective | Processing on an individual basis. Only fresh or frozen samples required | [29,75] | |
| 2-D Gel Analysis | Protein | Medium | resolution of complex protein mixtures, identification of evolutionary relatedness | Low | Evolutionary inference of taxa; analysis of polypeptides | Dependency of polypeptides resolved and polymorphism on sample number | [76] | |
| Mass Spectrometry (MALDI-TOF) | Protein | High | Identification of known species; diagnosis of PR proteins | High | High taxonomic resolution | Lack of genomic sequence | [77] | |
| Serological Analysis | Antigen/Antibodies | Medium | Generation of antisera against nematodes | High | Requirement of a low amount of protein in some cases | Lack of cross-reactivity | [78] | |
| New Emerging and Image-Based | ||||||||
| Machine Learning/A.I. | Image, annotation, and algorithm | Medium | Detection of phenotypes | Low | Nematode taxonomy and quantification. Fast and accurate identification | Multiple stages and requirement of expertise | [79,80] | |
| Autofluorescence | Natural autofluorescenceof microorganisms | Medium | Utilization of natural autofluorescence of microorganisms to substitute traditional light microscopy | Low | Easy emission and excitation spectra studies | The sample should include the autofluorescent itself | [81] | |
| Second-generation high-throughput sequencing | DNA | High | Nematode taxonomy (both known and unknown species) and quantification | Low | Cost-effective, depending upon the sample size | Requirement of field expert | [82] | |
| Microfluidic PCR Technique | DNA | Medium | Nematode detection and identification of known and unknown | Low | High-throughput analysis; sensitivity, specificity, and cost-effective | Specific target requirement | Not applied yet to roundworms | |
| Microarrays | DNA | High | Detection and identification and analysis of multiple genes | High | Isolation of pathogen not required; high-density probes for better analysis | Expensive and time-consuming | [83] | |
5. Biochemical- and Protein-Based Methods of Identification
5.1. Analysis Based on Isozymes
5.2. Use of Two-Dimensional Gel (2-DGE) Analysis
5.3. Serological Analysis or Use of Antibodies
5.4. Use of Mass Spectrometry Analysis
6. Emerging Methods of Nematode Identification
6.1. Image-Based Analysis and Identification
6.1.1. Use of the Deep Learning Approach
6.1.2. Use of Autofluorescence
6.2. Use of DNA Microarrays
6.3. Use of the Microfluidic PCR Technique
6.4. Use of the High-Throughput System for Massive Identification
7. Nematode Repositories and Databases
7.1. NeMys
7.2. Helminth.net
7.3. Nematode.net
7.4. NemaPath
7.5. Helminth Control and Prevention (HelmCoP)
7.6. NEMBASE
7.7. WormBase
7.8. WormBook
7.9. USDA Nematode Collection
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abad, P.; Gouzy, J.; Aury, J.M.; Castagnone-Sereno, P.; Danchin, E.G.J.; Deleury, E.; Perfus-Barbeoch, L.; Anthouard, V.; Artiguenave, F.; Blok, V.C.; et al. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat. Biotechnol. 2008, 26, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Abebe, E.; Mekete, T.; Thomas, W.K. A critique of current methods in nematode taxonomy. Afr. J. Biotechnol. 2011, 10, 312–323. [Google Scholar]
- Sibert, J.R. Intertidal hyperbenthic populations in the Nanaimo Estuary. Mar. Biol. 1981, 64, 259–265. [Google Scholar] [CrossRef]
- Roeber, F.; Jex, A.R.; Gasser, R.B. Next-generation molecular-diagnostic tools for gastrointestinal nematodes of livestock, with an emphasis on small ruminants: A turning point? Adv. Parasitol. 2013, 83, 267–333. [Google Scholar]
- De Oliveira, C.M.G.; Monteiro, A.R.; Blok, V.C. Morphological and molecular diagnostics for plant-parasitic nematodes: Working together to get the Identification done. Trop. Plant Pathol. 2011, 36, 65–73. [Google Scholar]
- Blaxter, M.L.; De Lay, P.; Garey, J.R.; Liu, L.X.; Scheldeman, P.; Vierstraete, A.; Vanfleteren, A.; Vanfleteren, J.R.; Mackey, L.Y.; Dorris, M.; et al. A molecular evolutionary framework for the phylum Nematoda. Nature 1998, 392, 71–75. [Google Scholar] [CrossRef]
- Meneely, P.M.; Dahlberg, C.L.; Rose, J.K. Working with worms: Caenorhabditis elegans as a model organism. Curr. Protoc. Essent. Lab. Tech. 2019, 19, e35. [Google Scholar] [CrossRef] [Green Version]
- Ferri, E.; Barbuto, M.; Bain, O.; Galimberti, A.; Uni, S.; Guerrero, R.; Ferté, H.; Bandi, C.; Martin, C.; Casiraghi, M. Integrated taxonomy: Traditional approach and DNA barcoding for the identification of filarioid worms and related parasites (Nematoda). Front. Zool. 2009, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Hunt, D.; Zafar, H. Taxonomy, Identification, and principal species. Root-Knot Nematodes 2009, 1, 55–97. [Google Scholar] [CrossRef]
- Roeber, F.; Kahn, L. The specific diagnosis of gastrointestinal nematode infections in livestock: Larval culture technique, its limitations, and alternative DNA-based approaches. Vet. Parasitol. 2014, 205, 619–628. [Google Scholar] [CrossRef]
- Gasser, R.B. Molecular tools—Advances, opportunities, and prospects. Vet. Parasitol. 2006, 136, 69–89. [Google Scholar] [CrossRef]
- Huette, R.N.; Golden, A.M. Nathan augustus COBB: The Father of Nematology in the United States. Annu. Rev. Phytopathol. 1991, 29, 15–26. [Google Scholar] [CrossRef]
- Dayrat, B. Towards integrative taxonomy. Biol. J. Linn. Soc. 2005, 85, 407–415. [Google Scholar] [CrossRef]
- Floyd, R.; Abebe, E.; Papert, A.; Blaxter, M. Molecular barcodes for soil nematode identification. Mol. Ecol. 2002, 11, 839–850. [Google Scholar] [CrossRef]
- Hunt, D.J.; Palomares-Rius, J.; Manzanilla-López, R.H. Identification, Morphology and Biology of Plant Parasitic Nematodes. In Plant Parasitic Nematodes in Subtropical and Tropical Agriculture, 3rd ed.; Sikora, R.A., Coyne, D., Hallmann, J., Timper, P., Eds.; CABI: Boston, MA, USA, 2018; Volume 10, pp. 20–61. [Google Scholar] [CrossRef]
- Poveda, J.; Abril-Urias, P.; Escobar, C. Biological Control of Plant-Parasitic Nematodes by Filamentous Fungi Inducers of Resistance: Trichoderma, Mycorrhizal and Endophytic Fungi. Front. Microbiol. 2020, 11, 992. [Google Scholar] [CrossRef]
- Evangelina, G.L.; Sánchez-Puerta, M.V. Characterization of a Root-Knot Nematode Population of Meloidogyne arenaria from Tupungato (Mendoza, Argentina). J. Nematol. 2012, 3, 291–301. [Google Scholar]
- Mir, R.A.; Bhat, K.A.; Rashid, G.; Ebinezer, L.B.; Masi, A.; Rakwal, R.; Shah, A.A.; Zargar, S.M. DNA barcoding: A way forward to obtain deep insights about the realistic diversity of living organisms. Nucleus 2021, 2, 157–165. [Google Scholar] [CrossRef]
- Ahmed, M.; Sapp, M.; Prior, T.; Karssen, G.; Back, M. Nematode taxonomy: From morphology to metabarcoding. Soil Discuss. 2015, 2, 1175–1220. [Google Scholar]
- Blok, V.C. Achievements in and future prospects for molecular diagnostics of plant-parasitic nematodes. Can. J. Plant Path. 2005, 2, 176–185. [Google Scholar] [CrossRef]
- Reslova, N.; Skorpikova, L.; Kyrianova, I.A.; Vadlejch, J.; Höglund, J.; Skuce, P.; Kasny, M. The identification and semi-quantitative assessment of gastrointestinal nematodes in faecal samples using multiplex real-time PCR assays. Parasit. Vectors 2021, 9, 391. [Google Scholar] [CrossRef]
- Ibrahim, I.; Handoo, Z.A.; Basyony, A.B. The cyst nematodes Heterodera and Globodera species in Egypt. Pak. J. Nematol. 2017, 2, 151–154. [Google Scholar] [CrossRef]
- Madani, M.; Subbotin, S.A.; Moens, M. Quantitative detection of the potato cyst nematode, Globodera pallida, and the beet cyst nematode, Heterodera schachtii, using Real-Time PCR with SYBR green I dye. Mol. Cell Probes 2005, 2, 81–86. [Google Scholar] [CrossRef]
- Shah, A.A.; Mir, R.A. Role of DNA-based markers in nematode taxonomy: A review. Int. J. Nematol. 2015, 2, 208–214. [Google Scholar]
- Ndao, M. Diagnosis of parasitic diseases: Old and new approaches. Interdiscip. Perspect. Infect. Dis. 2009, 2009, 278246. [Google Scholar] [CrossRef]
- Handoo, Z.A.; Carta, L.K.; Skantar, A.M. Taxonomy, Morphology, and Phylogenetics of Coffee-Associated Root-Lesion Nematodes, Pratylenchus spp. In Plant-Parasitic Nematodes of Coffee; Souza, R.M., Ed.; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Gasser, R.B.; Bott, N.J.; Chilton, N.B.; Hunt, P.; Beveridge, I. Toward practical, DNA based diagnostic methods for parasitic nematodes of livestock—Bionomic and biotechnological implications. Biotechnol. Adv. 2008, 26, 325–334. [Google Scholar] [CrossRef]
- Mattiucci, S.; Nascetti, G. Chapter 2: Advances and Trends in the Molecular Systematics of Anisakid Nematodes, with Implications for their Evolutionary Ecology and Host-Parasite Co-Evolutionary Processes. Adv. Parasitol. 2008, 66, 47–148. [Google Scholar] [CrossRef] [PubMed]
- Thevenoux, R.; Folcher, L.; Esquibet, M.; Fouville, D.; Montarry, J.; Grenier, E. The hidden diversity of the potato cyst nematode Globodera pallida in the south of Peru. Evol. Appl. 2020, 13, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, R.M.D.G.; Correa, V.R.; Almeida, M.R.A.; Gomes, A.C.M.; Deimi, A.M.; Castagnone-Sereno, P.; Karssen, G. Meloidogyne lucin. sp. (Nematoda: Meloidogynidae), a root-knot nematode parasitizing different crops in Brazil, Chile, and Iran. Nematology 2004, 16, 289–301. [Google Scholar] [CrossRef]
- Seesao, Y.; Audebert, C.; Verrez-Bagnis, V.; Merlin, S.; Jérôme, M.; Viscogliosi, E.; Dei-Cas, E.; Aliouat-Denis, C.M.; Gay, M. Monitoring of four DNA extraction methods upstream of high-throughput sequencing of Anisakidae nematodes. J. Microbiol. Methods 2014, 102, 69–72. [Google Scholar] [CrossRef]
- Dawkins, H.J.S.; Spencer, T.L. The isolation of nucleic acid from nematodes requires an understanding of the parasite and its cuticular structure. Parasitol. Today 1989, 5, 73–76. [Google Scholar] [CrossRef]
- Karanastasi, E.; Decraemer, W.; Zheng, J.; De Almeida, M.T.M.; Brown, D.J. Interspecific differences in the fine structure of the body cuticle of Trichodoridae Thorne, 1935 (Nematoda: Diphtherophorina) and review of anchoring structures of the epidermis. Nematology 2001, 3, 525–533. [Google Scholar]
- Castagnone-Sereno, P. Molecular tools for diagnosis. In Genomics and Molecular Genetics of Plant Nematode Interactions, 1st ed.; Jones, J., Gheisen, G., Fenoll, C., Eds.; Springer: New York, NY, USA, 2011; pp. 443–464. [Google Scholar]
- Anderson, R.C. Nematode Parasites of Vertebrates: Their Development and Transmission; C.A.B.I. U.S.D.A.: Wallingford, UK, 2000; Volume 8. [Google Scholar] [CrossRef]
- Bogale, M.; Baniya, A.; Di Gennaro, P. Nematode Identification Techniques, and Recent Advances. Plants 2020, 24, 1260. [Google Scholar] [CrossRef] [PubMed]
- Seesao, Y.; Gay, M.; Merlin, S.; Viscogliosi, E.; Aliouat-Denis, C.M.; Audebert, C.A. review of methods for nematode identification. J. Microbiol. Methods 2017, 138, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Holterman, M.M.; Oggenfuss, M.; Frey, J.E.; Kiewnik, S. Evaluation of high-resolution melting curve analysis as a new tool for root-knot nematode diagnostics. Phytopathology 2012, 160, 59–66. [Google Scholar] [CrossRef]
- Blok, V.C.; Powers, T.O. Biochemical and Molecular Identification; Root-Knot Nematodes by R.N. Perry, M. Moens, and J. L. Starr; C.A.B.I. Flanders Research Institute for agriculture, Fisheries and Food: Ghent, Belgium, 2009; pp. 98–118. [Google Scholar]
- Subbotin, S.A.; Halford, P.D.; Warry, A.; Perry, R.N. Variations in ribosomal DNA sequences and phylogeny of Globodera parasitizing solanaceous plantas. Nematology 2000, 2, 591–604. [Google Scholar] [CrossRef] [Green Version]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific Enzymatic Amplification of DNA in vitro: The Polymerase Chain Reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Pontes, T.; D’Amelio, S.; Costa, G.; Paggi. Molecular characterization of larval anisakid nematodes from marine fishes of Madeira by a PCR-based approach, with evidence for a new species. J. Parasitol. 2005, 91, 1430–1434. [Google Scholar] [CrossRef]
- Pineda, O.; Bonierbale, M.W.; Plaisted, R.L.; Brodie, B.B.; Tanksley, S.D. Identification of RFLP markers linked to the H1 gene conferring resistance to the potato cyst nematode Globodera rostochiensis. Genome 1993, 36, 152–156. [Google Scholar] [CrossRef]
- Cameron, M.; Levy, P.; Nutman, T.; Vanamala, C.; Narayanan, P.; Rajan, T. Use of restriction fragment length polymorphisms (RFLPs) to distinguish between nematodes of pathogenic significance. Parasitology 1988, 96, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Sedlák, P.; Melounová, M.; Skupinová, S.; Vejl, P.; Domkářová, J. Study of European and Czech populations of potato cyst nematodes (Globodera rostochiensis and G. pallida) by RAPD method. Plant Soil Environ. 2004, 50, 10. [Google Scholar] [CrossRef] [Green Version]
- Correa, V.R.; Mattos, V.S.; Almeida, M.R.A.; Santos, M.F.A.; Tigano, M.S.; Castagnone-Sereno, P.; Carneiro, R.M.D.G. Genetic diversity of the root-knot nematode Meloidogyne ethiopica and development of a species-specific SCAR marker for its diagnosis. Plant Pathol. 2014, 63, 476–483. [Google Scholar] [CrossRef] [Green Version]
- Castagnone-Sereno, P.; Vanlerberghe-Masutti, F.; Leroy, F. Genetic polymorphism between and within Meloidogyne species detected with RAPD markers. Genome Natl. Res. Counc. Can. Génome Cons. Natl. Rech. Can. 1995, 37, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Randig, O.; Leroy, F.; Castagnone-Sereno, P.R.A.P.D. Characterization of Single Females of the Root-knot Nematodes, Meloidogyne spp. Eur. J. Plant Pathol. 2001, 107, 639–643. [Google Scholar] [CrossRef]
- Höglund, J.; Engström, A.; Morrison, D.; Mattsson, J. Genetic diversity assessed by amplified fragment length polymorphism analysis of the parasitic nematode Dictyocaulus viviparus the lungworm of cattle. Int. J. Parasitol. 2004, 34, 475–484. [Google Scholar] [CrossRef]
- Li, Y.; Lawrence, G.W.; Lu, S.; Balbalian, C.; Klink, V.P. Quantitative field testing Heterodera glycines from metagenomic DNA samples isolated directly from soil under agronomic production. PLoS ONE 2014, 9, e89887. [Google Scholar] [CrossRef] [Green Version]
- Marché, L.; Valette, S.; Grenier, E.; Mugniéry, D. Intra-species DNA polymorphism in the tobacco cyst-nematode complex (Globodera tabacum) using A.F.L.P. Genome 2001, 44, 941–946. [Google Scholar]
- Fang, W.; Xu, S.; Zhang, S.; Wang, Y.; Chen, X.; Luo, D. Multiple primer PCR for the Identification of anisakid nematodes from Taiwan Strait. Exp. Parasitol. 2010, 124, 197–201. [Google Scholar] [CrossRef]
- Umehara, A.; Kawakami, Y.; Araki, J.; Uchida, A. Multiplex PCR for the Identification of Anisakis simplex Sensu stricto, Anisakis pegreffii, and the other anisakid nematodes. Parasitol. Int. 2008, 57, 49–53. [Google Scholar] [CrossRef]
- Sint, D.; Raso, L.; Traugott, M. Advances in multiplex PCR: Balancing primer efficiencies and improving detection success. Methods Ecol Evol. 2012, 3, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Mossali, C.; Palermo, S.; Capra, E.; Piccolo, G.; Botti, S.; Bandi, C.; D’Amelio, S.; Giuffra, E. Sensitive detection and quantification of anisakid parasite residues in food products. Foodborne Pathog. Dis. 2010, 7, 391–397. [Google Scholar] [CrossRef]
- Fang, W.; Liu, F.; Zhang, S.; Lin, J.; Xu, S.; Luo, D. Anisakis pegreffii: A quantitative fluorescence PCR assay for detection in situ. Exp. Parasitol. 2011, 127, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Stirling, G.; Griffin, D.; Ophel-Keller, K.; McKay, A.; Hartley, D.; Currar, J.; Stirling, A.; Monsour, C.; Winch, J.; Hardie, B. Combining an initial risk assessment process with DNA assays to improve prediction of soilborne diseases caused by root-knot nematode (Meloidogyne spp.) and Fusarium oxysporum f. sp. lycopersici in the Queensland tomato industry. Australas. Plant Pathol. 2004, 33, 285–293. [Google Scholar] [CrossRef]
- Amiri, S.; Subottin, S.A.; Moens, M. Identification of the beet cyst nematode Heterodera schachtii by PCR. Eur. J. Plant Pathol. 2002, 108, 497–506. [Google Scholar] [CrossRef]
- Sapkota, R.; Skantar, A.M.; Nicolaisen, M. A TaqMan real-time PCR assay for detection of Meloidogyne hapla in root galls and soil. Nematology 2016, 18, 147–154. [Google Scholar] [CrossRef]
- Huang, D.; Yan, G.; Gudmestad, N.; Skantar, A. Quantification of Paratrichodorus allius in DNA extracted from soil using TaqMan Probe and S.Y.B.R. Green real-time PCR assays. Nematology 2017, 19, 987–1001. [Google Scholar] [CrossRef]
- Toumi, F.; Waeyenberge, L.; Viaene, N.; Dababat, A.; Nicol, J.M.; Ogbonnaya, F.; Moens, M. Development of two species-specific primer sets to detect the cereal cyst nematodes Heterodera avenae and Heterodera filipjevi. Eur. J. Plant Pathol. 2013, 136, 613–624. [Google Scholar] [CrossRef]
- Van Megen, H.; Van Den Elsen, S.; Holterman, M.; Karssen, G.; Mooyman, P.; Bongers, T.; Holovachov, O.; Bakker, J.; Helder, J. A phylogenetic tree of nematodes based on about 1200 full-length small subunit ribosomal DNA sequences. Nematology 2009, 11, 927–950. [Google Scholar] [CrossRef]
- Hadziavdic, K.; Lekang, K.; Lanzen, A.; Jonassen, I.; Thompson, E.M.; Troedsson, C. Characterization of the 18s rRNA gene for designing universal eukaryote specific primers. PLoS ONE 2014, 9, e87624. [Google Scholar] [CrossRef] [Green Version]
- DeSalle, R.; Egan, M.G.; Siddall, M. The unholy trinity: Taxonomy, species delimitation, and DNA barcoding. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1905–1916. [Google Scholar] [CrossRef] [Green Version]
- Bu, Y.; Niu, H.; Zhang, L. Phylogenetic analysis of the genus Cylicocyclus (Nematoda: Strongylidae) based on nuclear ribosomal sequence data. Acta Parasitol. 2013, 58, 167–173. [Google Scholar] [CrossRef]
- Félix, M.A.; Braendle, C.; Cutter, A.D. A streamlined system for species diagnosis in Caenorhabditis (Nematoda: Rhabditidae) with name designations for 15 distinct biological species. PLoS ONE 2014, 9, e94723. [Google Scholar] [CrossRef] [PubMed]
- Zajac, A.M. Gastrointestinal nematodes of small ruminants: Life cycle, anthelmintics, and diagnosis. Vet. Clin. N. Am. Food Anim. Pract. 2006, 22, 529–541. [Google Scholar] [CrossRef] [PubMed]
- McLeod, R.S. Costs of major parasites to the Australian livestock industries. Int. J. Parasitol. 1995, 25, 1363–1367. [Google Scholar] [CrossRef]
- Zarlenga, D.S.; Hoberg, E.P.; Stringfellow, F.; Lichtenfels, J.R. Comparisons of two polymorphic species of Ostertagia and phylogenetic relationships within the Ostertagiinae (Nematoda: Trichostrongyloidea) inferred from ribosomal DNA repeat and mitochondrial DNA sequences. J. Parasitol. 2001, 84, 806–812. [Google Scholar] [CrossRef]
- Chilton, N.B.; Newton, L.A.; Beveridge, I.; Gasser, R.B. Evolutionary relationships of trichostrongyloid nematodes (Strongylida) inferred from ribosomal DNA sequence data. Mol. Phylogenet. Evol. 2001, 19, 367–386. [Google Scholar] [CrossRef] [PubMed]
- DellAnno, A.; Carugati, L.; Corinaldesi, C.; Riccioni, G.; Danovaro, R. Unveiling the Biodiversity of Deep-Sea Nematodes through Metabarcoding: Are We Ready to Bypass the Classical Taxonomy? PLoS ONE 2015, 10, e0144928. [Google Scholar] [CrossRef] [PubMed]
- Haenel, Q.; Holovachov, O.; Jondelius, U.; Sundberg, P.; Bourlat, S. NGS-based biodiversity, and community structure analysis of meiofaunal eukaryotes in shell sand from Hållö island, Smögen, and soft mud from Gullmarn Fjord. Sweden. Biodivers. Data J. 2017, 5, e12731. [Google Scholar] [CrossRef] [Green Version]
- Leasi, F.; Sevigny, J.L.; Laflamme, E.M.; Artois, T.; Curini-Galletti, M.; de Jesus Navarrete, A.; Thomas, W.K. Biodiversity estimates and ecological interpretations of meiofaunal communities are biased by the taxonomic approach. Commun. Biol. 2018, 1, 112. [Google Scholar] [CrossRef] [Green Version]
- Holovachov, O. Metabarcoding of marine nematodes—Evaluation of similarity scores used in alignment-based taxonomy assignment approach. Biodivers. Data J. 2016, 4, e10647. [Google Scholar] [CrossRef]
- Macheriotou, L.; Guilini, K.; Bezerra, T.N.; Tytgat, B.; Nguyen, D.T.; Phuong Nguyen, T.X.; Derycke, S. Metabarcoding free-living marine nematodes using curated 18S and CO1 reference sequence databases for species-level taxonomic assignments. Ecol. Evol. 2019, 9, 1211–1226. [Google Scholar] [CrossRef] [Green Version]
- Holovachov, O.; Haenel, Q.; Bourlat, S.J.; Jondelius, U. Taxonomy assignment approach determines the efficiency of identification of O.T.U.s in marine nematodes. R. Soc. Open Sci. 2017, 4, 170315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, J.; Kleinbölting, N.; Traunspurger, W. Comparison of morphological, DNA barcoding, and metabarcoding characterizations of freshwater nematode communities. Ecol. Evol. 2020, 10, 2885–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillou, L.; Bachar, D.; Audic, S.; Bass, D.; Berney, C.; Bittner, L.; Boutte, C.; Burgaud, G.; de Vargas, C.; Decelle, J.; et al. The Protist Ribosomal Reference database (PR2): A catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2013, 41, 597–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, T.J.; De Santiago, A.; Schuelke, T.; Hardy, S.M.; Bik, H.M. The impact of intragenomic rRNA variation on metabarcoding derived diversity estimates: A case study from marine nematodes. Environ. DNA 2020, 2, 519–534. [Google Scholar] [CrossRef] [Green Version]
- Davey, M.L.; Utaaker, K.S.; Fossøy, F. Characterizing parasitic nematode faunas in faeces and soil using DNA metabarcoding. Parasites Vectors 2021, 14, 422. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581. [Google Scholar] [CrossRef] [Green Version]
- Conole, J.C.; Chilton, N.B.; Järvis, T.; Gasser, R.B. Intraspecific and interspecific variation in the second internal transcribed spacer sequence for Metastrongylus (Nematoda: Metastrongyloidea) detected by high-resolution PCR-linked restriction fragment length polymorphism. Int. J. Parasitol. 1999, 29, 1935–1940. [Google Scholar] [CrossRef]
- Qin, L.; Overmars, H.; Helder, J.; Popeijus, H.; van der Voort, J.R.; Groenink, W.; van Koert, P.; Schots, A.; Bakker, J.; Smant, G. An efficient cDNA-AFLP-based strategy for the Identification of putative pathogenicity factors from the potato cyst nematode Globodera rostochiensis. Mol. Plant-Microbe Interact. 2000, 13, 830–836. [Google Scholar] [CrossRef] [Green Version]
- Pedram, M.; Pourjam, E.; Atighi, M.R.; Panahandeh, Y. Further studies on soil nematode fauna in northwestern Iran with the description of one new species. J. Nematol. 2015, 47, 148. [Google Scholar]
- Valentini, A.; Mattiucci, S.; Bondanelli, P. Genetic relationships among Anisakis species (Nematoda: Anisakidae) inferred from mitochondrial Cox2 sequences, and comparison with allozyme data. J. Parasitol. 2006, 92, 156–166. [Google Scholar] [CrossRef]
- Navas, A.; López, J.A.; Espárrago, G.; Camafeita, E.; Albar, J.P. Protein variability in Meloidogyne spp. (Nematoda: Meloidogynidae) revealed by two-dimensional gel electrophoresis and mass spectrometry. J. Proteome Res. 2002, 1, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Millares, P.; LaCourse, E.J.; Perally, S.; Ward, D.A.; Prescott, M.C.; Hodgkinson, J.E.; Brophy, P.M.; Rees, H.H. Proteomic profiling and protein identification by MALDI-TOF mass spectrometry in unsequenced parasitic nematodes. PLoS ONE 2012, 7, e33590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schots, A.; Hermsen, T.; Schouten, S.; Gommers, F.J.; Egberts, E. Serological differentiation of the potato-cyst nematodes Globdera pallida and G. rostochiensis: II. Preparation and characterization of species-specific monoclonal antibodies. Hybridoma 1989, 8, 401–413. [Google Scholar] [CrossRef]
- Akintayo, A.; Tylka, G.L.; Singh, A.K.; Ganapathysubramanian, B.; Singh, A.; Sarkar, S. A deep learning framework to discern and count microscopic nematode eggs. Sci. Rep. 2018, 8, 9145. [Google Scholar] [CrossRef] [PubMed]
- Hakim, A.; Mor, Y.; Toker, I.A.; Levine, A.; Neuhof, M.; Markovitz, Y.; Rechavi, O. WorMachine: Machine learning-based phenotypic analysis tool for worms. BMC Biol. 2018, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qazi, F.; Khalid, A.; Poddar, A.; Tetienne, J.P.; Nadarajah, A.; Aburto-Medina, A.; Shahsavari, E.; Shukla, R.; Prawer, S.; Ball, A.S.; et al. Real-time detection and Identification of nematode eggs genus and species through optical imaging. Sci. Rep. 2020, 10, 7219. [Google Scholar] [CrossRef]
- Eves-van den Akker, S.; Lilley, C.J.; Reid, A.; Pickup, J.; Anderson, E.; Cock, P.J.; Blaxter, M.; Urwin, P.E.; Jones, J.T.; Blok, V.C. A metagenetic approach to determine the diversity and distribution of cyst nematodes at the level of the country, the field and the individual. Mol. Ecol. 2015, 24, 5842–5851. [Google Scholar] [CrossRef] [Green Version]
- Golden, T.; Hubbard, A.; Melov, S. Microarray analysis of variation in individual aging C. elegans: Approaches and challenges. Exp. Gerontol. 2006, 41, 1040–1045. [Google Scholar] [CrossRef]
- Esbenshade, P.R.; Triantaphyllou, A.C. Isozyme phenotypes for the Identification of Meloidogyne species. J. Nematol. 1990, 22, 10–15. [Google Scholar]
- Bird, A.F. Serological studies on the plant-parasitic nematode, Meloidogyne javanica. Exp. Parasitol. 1964, 15, 350–360. [Google Scholar] [CrossRef]
- Lee, S.H. Attempts to use immunodiffusion for species identification of Meloidogyne (Abstr.). Nematologica 1965, 11, 41. [Google Scholar]
- Misaghi, I.; McClure, M.A. Antigenic Relationship of Meloidogyne incognita, M. javanica, and M. arenaria. Phytopathology 1974, 64, 698–701. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.D.; Chen, Y.J.J.; Wu, J.; Chaudhuri, S.; Hsiao, Y.C.; Schneider, K.; Hoi, K.H.; Lin, Z.; Guerrero, S.; Jaiswal, B.S.; et al. Massively parallel single-cell B-cell receptor sequencing enables rapid discovery of diverse antigen-reactive antibodies. Commun. Biol. 2019, 2, 304. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Babalola, O.O.; Tak, H.I. Potential of MALDI-ToF mass spectrometry as a rapid detection technique in plant pathology: Identification of plant-associated microorganisms. Anal. Bioanal. Chem. 2012, 404, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.R.; Vanstone, V.A.; Jones, M.G.K. A novel approach to identify plant-parasitic nematodes using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Biron, D.G.; Joly, C.; Marché, L.; Galéotti, N.; Calcagno, V.; Schmidt-Rhaesa, A.; Renault, L.; Thomas, F. First analysis of the proteome in two nematomorph species, Paragordius tricuspidatus (Chordodidae) and Spinochordodes tellinii (Spinochordodidae). Infect. Genet. Evol. 2005, 5, 167–175. [Google Scholar] [CrossRef]
- Monis, P.T.; Giglio, S.; Keegan, A.R.; Thompson, R.A. Emerging technologies for the detection and genetic characterization of protozoan parasites. Trends Parasitol. 2005, 21, 340–346. [Google Scholar] [CrossRef]
- Hino, A.; Maruyama, H.; Kikuchi, T. A novel method to assess the biodiversity of parasites using 18S rDNA Illumina sequencing; parasitome analysis method. Parasitol. Int. Online 2016, 65, 572–575. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y.Y.; Liu, B.C.; Zhang, L.P. Morphological and molecular evidence for a new species of the genus Raphidascaris (Nematoda: Anisakidae) from marine fishes from the South China Sea. Parasitol. Res. 2012, 110, 1473–1479. [Google Scholar] [CrossRef]
- Ristau, K.; Steinfartz, S.; Traunspurger, W. First evidence of cryptic species diversity and significant population structure in a widespread freshwater nematode morphospecies (Tobrilus gracilis). Mol. Ecol. 2013, 22, 4562–4575. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next-generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatta, H.; Goldys, E.M.; Learmonth, R.P. Use fluorescence spectroscopy to differentiate yeast and bacterial cells. Appl. Microbiol. Biotechnol. 2006, 71, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, A.; Singh, S. Progress of science from microscopy to microarrays (part 1): Diagnosis of parasitic diseases. J. Lab. Physicians 2009, 1, 2. [Google Scholar] [CrossRef]
- Klink, V.P.; Hosseini, P.; Matsye, P.D.; Alkharouf, N.W.; Matthews, B.F. Syncytium gene expression in Glycine max [PI 88788] roots undergoing a resistant reaction to the parasitic nematode Heterodera glycines. Plant Physiol. Biochem. 2010, 48, 176–193. [Google Scholar] [CrossRef]
- Ahmed, M.; Singh, M.; Bera, A.; Bandyopadhyay, S.; Bhattacharya, D. Molecular basis for Identification of species/isolates of gastrointestinal nematode parasites. Asian Pac. J. Trop. Med. 2011, 4, 589–593. [Google Scholar] [CrossRef] [Green Version]
- Hudecova, I. Digital PCR analysis of circulating nucleic acids. Clin. Biochem. 2015, 48, 948–956. [Google Scholar] [CrossRef]
- Baker, M. Digital PCR hits its stride. Nat. Methods 2012, 9, 541–544. [Google Scholar] [CrossRef]
- Zhu, Z.; Jenkins, G.; Zhang, W.; Zhang, M.; Guan, Z.; Yang, C.J. Single-molecule emulsion PCR in microfluidic droplets. Anal. Bioanal. Chem. 2012, 403, 2127–2143. [Google Scholar] [CrossRef] [Green Version]
- Eastburn, D.J.; Sciambi, A.; Abate, A.R. Picoinjection enables digital detection of RNA with droplet rt-PCR. PLoS ONE 2013, 26, e62961. [Google Scholar] [CrossRef] [Green Version]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Kirschberg, M. How many 16S-based studies should be included in a metagenomic conference? It may be a matter of etymology. FEMS Microbiol. Lett. 2014, 351, 145–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porazinska, D.L.; Giblin-Davis, R.M.; Faller, L.; Farmerie, W.; Kanzaki, N.; Morris, K.; Powers, T.O.; Tucker, A.E.; Sung, W.; Thomas, W.K. Evaluating high-throughput sequencing as a method for metagenomic analysis of nematode diversity. Mol. Ecol. Resour. 2009, 9, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Darby, B.; Todd, T.C.; Herman, M.A. High-throughput amplicon sequencing of rRNA genes requires a copy number correction to accurately reflect the effects of management practices on soil nematode community structure. Mol. Ecol. 2013, 22, 5456–5471. [Google Scholar] [CrossRef] [Green Version]
- Audebert, C.; Hot, D.; Lemoine, Y.; Caboche, S. Le séquençage haut-débit-vers un diagnostic basé sur la séquence complète du génome de l’agent infectieux. Méd. Sci. 2014, 30, 1144–1151. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhat, K.A.; Mir, R.A.; Farooq, A.; Manzoor, M.; Hami, A.; Allie, K.A.; Wani, S.M.; Khan, M.N.; Sayyed, R.Z.; Poczai, P.; et al. Advances in Nematode Identification: A Journey from Fundamentals to Evolutionary Aspects. Diversity 2022, 14, 536. https://doi.org/10.3390/d14070536
Bhat KA, Mir RA, Farooq A, Manzoor M, Hami A, Allie KA, Wani SM, Khan MN, Sayyed RZ, Poczai P, et al. Advances in Nematode Identification: A Journey from Fundamentals to Evolutionary Aspects. Diversity. 2022; 14(7):536. https://doi.org/10.3390/d14070536
Chicago/Turabian StyleBhat, Kaisar Ahmad, Rakeeb Ahmad Mir, Asmat Farooq, Madhiya Manzoor, Ammarah Hami, Kaisar Ahmad Allie, Shaheen Majeed Wani, M. N. Khan, R. Z. Sayyed, Peter Poczai, and et al. 2022. "Advances in Nematode Identification: A Journey from Fundamentals to Evolutionary Aspects" Diversity 14, no. 7: 536. https://doi.org/10.3390/d14070536
APA StyleBhat, K. A., Mir, R. A., Farooq, A., Manzoor, M., Hami, A., Allie, K. A., Wani, S. M., Khan, M. N., Sayyed, R. Z., Poczai, P., Almalki, W. H., Zargar, S. M., & Shah, A. A. (2022). Advances in Nematode Identification: A Journey from Fundamentals to Evolutionary Aspects. Diversity, 14(7), 536. https://doi.org/10.3390/d14070536