
Development of EST-SSR Markers Related to Polyphyllin Biosynthesis Reveals Genetic Diversity and Population Structure in Paris polyphylla


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. EST-SSR Identification and Marker Development
2.3. Marker Validation
2.4. Data Analysis
3. Results
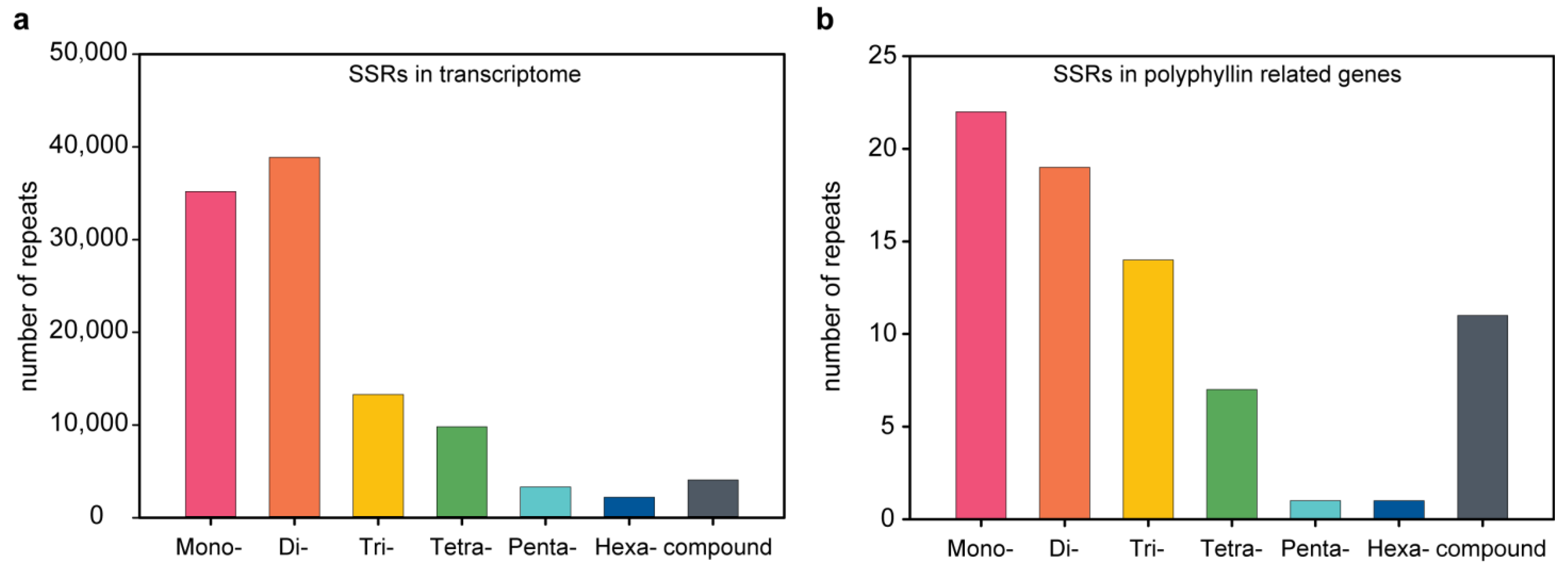
3.1. EST-SSR Identification
3.2. Development of EST-SSR Makers
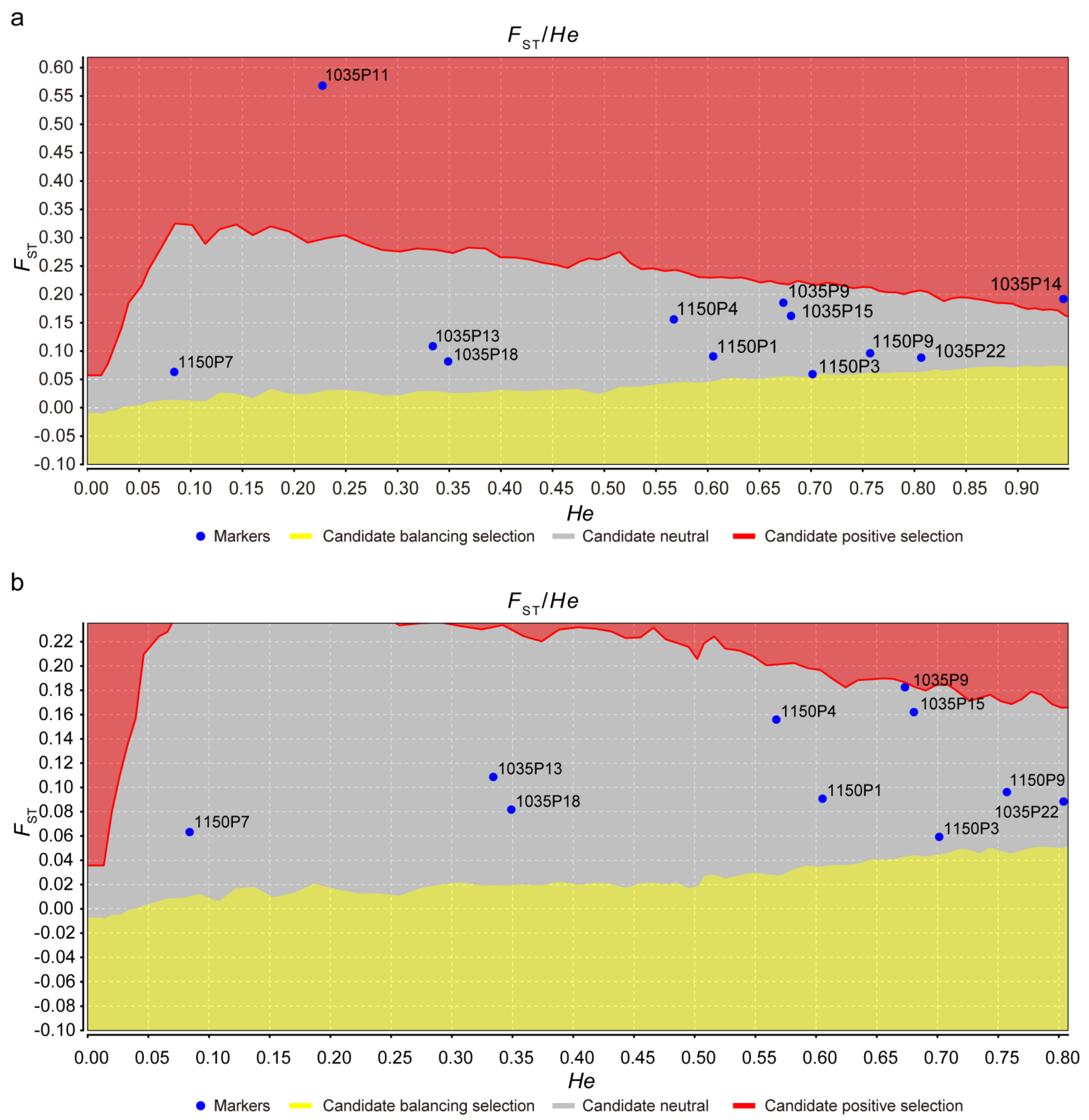
3.3. Polymorphism Analysis of SSR Loci
3.4. Genetic Diversity and Genetic Variation of Populations
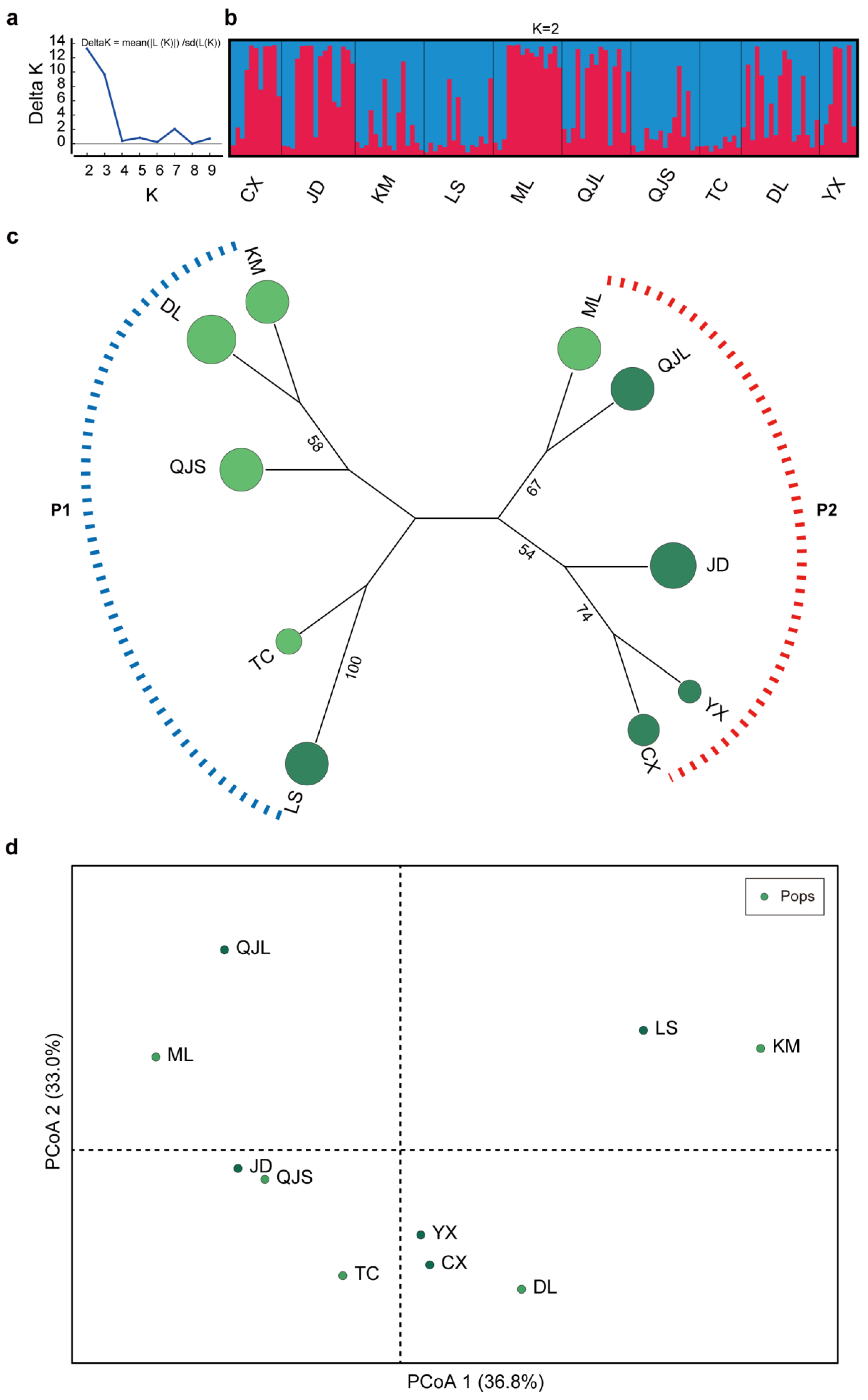
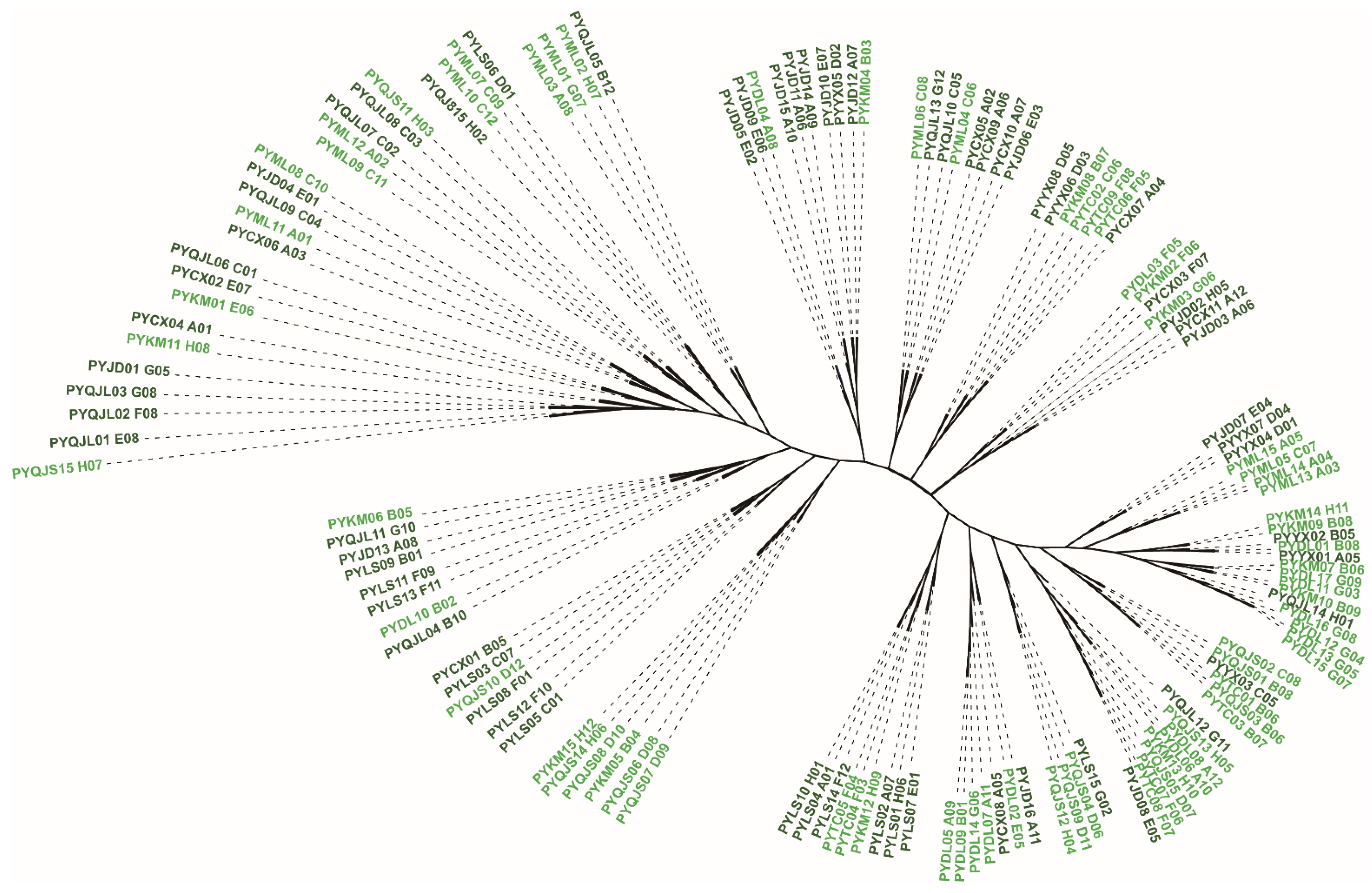
3.5. Genetic Structure and Population Clustering
4. Discussion
4.1. SSR Frequency and Distribution
4.2. Marker Polymorphism
4.3. Relationships in the Germplasm Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, L. The Genus Paris Plants; Science Press: Beijing, China, 2008; p. 33. [Google Scholar]
- Pei, Y.F.; Zhang, Q.Z.; Wang, Y.Z. Application of Authentication Evaluation Techniques of Ethnobotanical Medicinal Plant Genus Paris: A Review. Crit. Rev. Anal. Chem. 2020, 50, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.G.; Zhao, Y.L.; Zhang, J.; Zuo, Z.T.; Zhang, Q.Z.; Wang, Y.Z. The traditional uses, phytochemistry, and pharmacological properties of Paris L. (Liliaceae): A review. J. Ethnopharmacol. 2021, 278, 114293. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.B.; Brinckmann, J.A.; Bi, Y.F.; Pei, S.J.; Schippmann, U.; Luo, P. Paris in the spring: A review of the trade, conservation and opportunities in the shift from wild harvest to cultivation of Paris polyphylla (Trilliaceae). J. Ethnopharmacol. 2018, 222, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.K.; Kalia, S.K.; Kaul, S.; Dalal, V.; Hemaprabha, G.; Selvi, A.; Pandit, A.; Singh, A.; Gaikwad, K.; Sharma, T.R.; et al. Informative genomic microsatellite markers for efficient genotyping applications in sugarcane. Theor. Appl. Genet. 2009, 118, 327–338. [Google Scholar] [CrossRef]
- Grover, A.; Sharma, P.C. Development and use of molecular markers: Past and present. Crit. Rev. Biotechnol. 2016, 36, 290–302. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Wang, H.; Li, D.Z.; Chen, S.F. Genetic diversity of Paris polyphylla var. yunnanensis, a traditional Chinese medicinal herb, detected by ISSR markers. Planta Med. 2007, 73, 1316–1321. [Google Scholar]
- Huang, Y.; Zhou, N.; Yang, M.; Shen, Y.X.; Zhang, D.Q. A comparative study of the population genetics of wild and cultivated populations of Paris polyphylla var. yunnanensis based on amplified fragment length polymorphism markers. Ecol. Evol. 2019, 9, 10707–10722. [Google Scholar]
- Zhao, X.P.; Zou, G.F.; Zhao, J.; Hu, L.Y.; Lan, Y.F.; He, J.L. Genetic relationships and diversity among populations of Paris polyphylla assessed using SCoT and SRAP markers. Physiol. Mol. Biol. Plants. 2020, 26, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, Y.; Zhao, Y.; Yang, S.; Udikeri, S.; Liu, T. De Novo Characterization of the Root Transcriptome and Development of EST-SSR Markers in Paris polyphylla Smith var. yunnanensis, an Endangered Medical Plant. J. Agric. Sci. Technol. 2016, 18, 437–452. [Google Scholar]
- Pellicer, J.; Kelly, L.J.; Leitch, I.J.; Zomlefer, W.B.; Fay, M.F. A universe of dwarfs and giants: Genome size and chromosome evolution in the monocot family Melanthiaceae. New Phytol. 2014, 201, 1484–1497. [Google Scholar] [CrossRef]
- Cota-Sanchez, J.H.; Remarchuk, K.; Ubayasena, K. Ready-to-use DNA extracted with a CTAB method adapted for herbarium specimens and mucilaginous plant tissue. Plant Mol. Biol. Rep. 2006, 24, 161–167. [Google Scholar] [CrossRef]
- Gao, X.Y.; Zhang, X.; Chen, W.; Li, J.; Yang, W.J.; Zhang, X.W.; Li, S.Y.; Liu, C.N. Transcriptome analysis of Paris polyphylla var. yunnanensis illuminates the biosynthesis and accumulation of steroidal saponins in rhizomes and leaves. Phytochemistry 2020, 178, 12460. [Google Scholar]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Antao, T.; Lopes, A.; Lopes, R.J.; Beja-Pereira, A.; Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a F(st)-outlier method. BMC Bioinform. 2008, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, M.A.; Nichols, R.A. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. B-Biol. Sci. 1996, 263, 1619–1626. [Google Scholar]
- Ohtani, M.; Kondo, T.; Tani, N.; Ueno, S.; Lee, L.S.; Ng, K.K.S.; Muhammad, N.; Finkeldey, R.; Na’iem, M.; Indrioko, S.; et al. Nuclear and chloroplast DNA phylogeography reveals Pleistocene divergence and subsequent secondary contact of two genetic lineages of the tropical rainforest tree species Shorea leprosula (Dipterocarpaceae) in South-East Asia. Mol. Ecol. 2013, 22, 2264–2279. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takezaki, N.; Nei, M.; Tamura, K. POPTREE2: Software for Constructing Population Trees from Allele Frequency Data and Computing Other Population Statistics with Windows Interface. Mol. Biol. Evol. 2010, 27, 747–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.J.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Wright, S. Evolution in Mendelian populations. Genetics 1931, 16, 97–159. [Google Scholar] [CrossRef]
- Bland, J.M.; Altman, D.G. Multiple Significance Tests—The Bonferroni Method. Br. Med. J. 1995, 310, 170. [Google Scholar] [CrossRef] [Green Version]
- Retief, J.D. Phylogenetic analysis using PHYLIP. Methods Mol. Biol. 2000, 132, 243–258. [Google Scholar]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Comput. Appl. Biosci. 1994, 10, 189–191. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree 1.4. 2 Software. Institute of Evolutionary Biology, Univ. Edinburgh. 2014. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 1 July 2022).
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a Genetic-Linkage Map in Man Using Restriction Fragment Length Polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Parida, S.K.; Kumar, K.A.R.; Dalal, V.; Singh, N.K.; Mohapatra, T. Unigene derived microsatellite markers for the cereal genomes. Theor. Appl. Genet. 2006, 112, 808–817. [Google Scholar] [CrossRef]
- Li, B.; Peng, L.; Sun, X.C.; Huang, W.J.; Wang, N.; He, Y.H.; Shi, X.B.; Liu, Y.R.; Zhang, P.; Yang, X.J.; et al. Organ-specific transcriptome sequencing and mining of genes involved in polyphyllin biosynthesis in Paris polyphylla. Ind. Crop. Prod. 2020, 156, 112775. [Google Scholar] [CrossRef]
- Von Stackelberg, M.; Rensing, S.A.; Reski, R. Identification of genic moss SSR markers and a comparative analysis of twenty-four algal and plant gene indices reveal species-specific rather than group-specific characteristics of microsatellites. BMC Plant Biol. 2006, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Hodel, R.G.J.; Gitzendanner, M.A.; Germain-Aubrey, C.C.; Liu, X.X.; Crowl, A.A.; Sun, M.; Landis, J.B.; Segovia-Salcedo, M.C.; Douglas, N.A.; Chen, S.C.; et al. A New Resource for the Development of SSR Markers: Millions of Loci from a Thousand Plant Transcriptomes. Appl. Plant Sci. 2016, 4, 1600024. [Google Scholar] [CrossRef]
- Wu, Q.C.; Zang, F.Q.; Xie, X.M.; Ma, Y.; Zheng, Y.Q.; Zang, D.K. Full-length transcriptome sequencing analysis and development of EST-SSR markers for the endangered species Populus wulianensis. Sci. Rep. 2020, 10, 16249. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, X.M.; Tang, T.; Wang, Y.D.; Wu, Y.H.; Luo, J.Y.; Wu, H.T.; Wang, Y.Q.; Zhang, J.; Ruan, R.W.; et al. Inflorescence Transcriptome Sequencing and Development of New EST-SSR Markers in Common Buckwheat (Fagopyrum esculentum). Plants 2022, 11, 742. [Google Scholar] [CrossRef] [PubMed]
- You, Y.N.; Huang, X.F.; Liu, H.B.; Cheng, T.; Zheng, X.F.; Diao, Y.; Bao, Z.Z.; Dong, C.; Ke, W.D.; Hu, Z.L. Leaf Transcriptome Analysis and Development of EST-SSR Markers in Arrowhead (Sagittaria trifolia L. var. Sinensis). Trop. Plant Biol. 2020, 13, 189–200. [Google Scholar] [CrossRef]
- Lawson, M.J.; Zhang, L.Q. Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biol. 2006, 7, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Nevo, E. Microsatellites within genes: Structure, function, and evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.Y.; Wang, H.; Chen, X.X.; Wang, P.; Gao, P.; Li, X.N.; Zhu, G.P. Microsatellite markers for assessing genetic diversity of the medicinal plant Paris polyphylla var. chinensis (Trilliaceae) . Genet. Mol. Res. 2012, 11, 1975–1980. [Google Scholar] [CrossRef]
- Aranzana, M.J.; Illa, E.; Howad, W.; Arus, P. A first insight into peach [Prunus persica (L.) Batsch] SNP variability. Tree Genet. Genomes 2012, 8, 1359–1369. [Google Scholar] [CrossRef] [Green Version]
- Menken, S.B.J.; Smit, E.; DenNijs, H.C.M. Genetical population structure in plants: Gene flow between diploid sexual and triploid asexual dandelions (Taraxacum section Ruderalia). Evolution 1995, 49, 1108–1118. [Google Scholar] [CrossRef]
- Callen, D.F.; Thompson, A.D.; Shen, Y.; Phillips, H.A.; Richards, R.I.; Mulley, J.C.; Sutherland, G.R. Incidence and Origin of Null Alleles in the (Ac)N Microsatellite Markers. Am. J. Hum. Genet. 1993, 52, 922–927. [Google Scholar]
- Song, Y.; Li, M.F.; Xu, J.; Zhao, Z.; Chen, N.Z. Polymorphic microsatellite markers in the traditional Chinese medicinal plant Paris polyphylla var. yunnanensis. Genet. Mol. Res. 2015, 14, 9939–9942. [Google Scholar] [CrossRef]
- Chen, Z.S.Z. Genetic diversity of Paris polyphylla var. yunnanensis by SSR marker. Chin. Tradit. Herb. Drugs 2017, 48, 1834–1838. [Google Scholar]
- Qi, J.J.; Zheng, N.; Zhang, B.; Sun, P.; Hu, S.N.; Xu, W.J.; Ma, Q.; Zhao, T.Z.; Zhou, L.L.; Qin, M.J.; et al. Mining genes involved in the stratification of Paris Polyphylla seeds using high-throughput embryo transcriptome sequencing. BMC Genomics 2013, 14, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgro, C.M.; Lowe, A.J.; Hoffmann, A.A. Building evolutionary resilience for conserving biodiversity under climate change. Evol. Appl. 2011, 4, 326–337. [Google Scholar] [CrossRef]
- Ren, Z.X.; Wang, H.; Bernhardt, P.; Li, D.Z. Insect Pollination and Self-Incompatibility in Edible and/or Medicinal Crops in Southwestern China, a Global Hotspot of Biodiversity. Am. J. Bot. 2014, 101, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, D.W.; Templeton, A.R. Correlation of pairwise genetic and geographic distance measures: Inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 1999, 53, 1898–1914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Q.; Zhou, N. Genetic diversity and population structure of the endangered conifer Taxus wallichiana var. mairei (Taxaceae) revealed by Simple Sequence Repeat (SSR) markers. Biochem. Syst. Ecol. 2013, 49, 107–114. [Google Scholar]
- Toro, M.A.; Caballero, A. Characterization and conservation of genetic diversity in subdivided populations. Philos. Trans. R. Soc. Lond. B-Biol. Sci. 2005, 360, 1367–1378. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
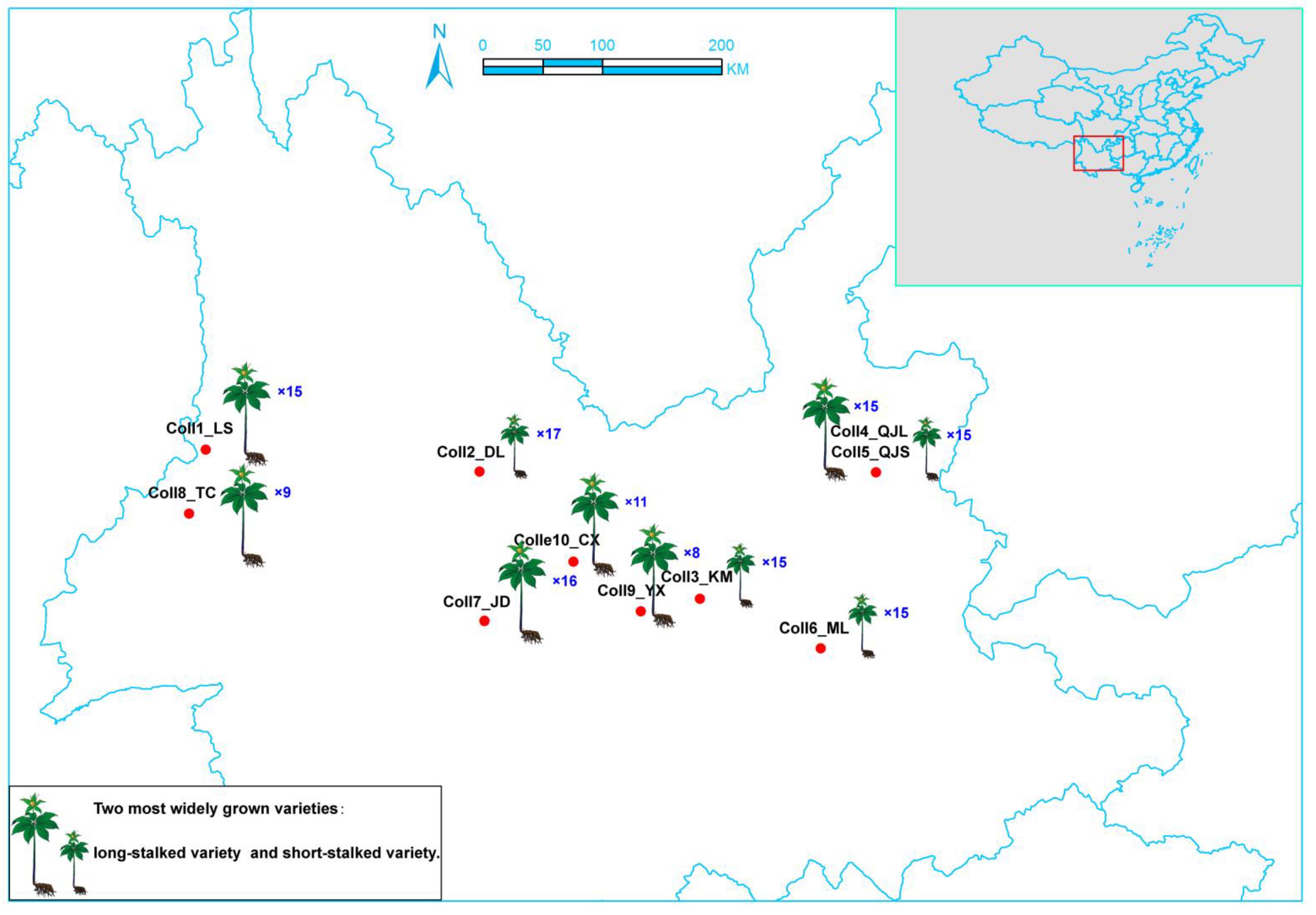
| Population | Sampling Location | City | Population Size | Varieties | Longitude (°) | Latitude (°) | Altitude (m) |
|---|---|---|---|---|---|---|---|
| Coll1_LS | Lushui | Nujiang | 15 | long stalk | 98.78 (E) | 25.91 (N) | 1446 |
| Coll2_DL | Xiangyun | Dali | 17 | short stalk | 100.84 (E) | 25.74 (N) | 1775 |
| Coll3_KM | Xishan | Kunming | 15 | short stalk | 102.50 (E) | 24.78 (N) | 2008 |
| Coll4_QJL | Zhanyi | Qujing | 15 | long stalk | 103.83 (E) | 25.73 (N) | 2204 |
| Coll5_QJS | Zhanyi | Qujing | 15 | short stalk | 103.83 (E) | 25.73 (N) | 2204 |
| Coll6_ML | Mile | Honghe | 15 | short stalk | 103.41(E) | 24.41 (N) | 1711 |
| Coll7_JD | Jingdong | Puer | 16 | long stalk | 100.88 (E) | 24.62 (N) | 2237 |
| Coll8_TC | Tengchong | Baoshan | 9 | short stalk | 98.65(E) | 25.42 (N) | 1850 |
| Coll9_YX | Yimen | Yuxi | 8 | long stalk | 102.06 (E) | 24.69 (N) | 1873 |
| Coll10_CX | Chuxiong | Chuxiong | 11 | long stalk | 101.49 (E) | 24.95 (N) | 1857 |
| Primers | Sequence (5’ to 3’) | Tm (°C) | SSR Type | Expected Product Size (bp) | 5’Modification | Gene Candidate | Polyphyllin Backbone Biosynthesis |
|---|---|---|---|---|---|---|---|
| STR1035-9F | CTATCGGAGAGTCTGACCCTAC | 55 | (GT)6 | 130 | 5’HEX | STE24 | downstream |
| STR1035-9R | GTAACCATTGATTTCCAGCTG | ||||||
| STR1035-11F | CAGAATAAAGACGGTGAATTAAAAT | 56 | (CGC)4 | 115 | 5’HEX | SMT2 | downstream |
| STR1035-11R | CCCATGCATATGATCCTCTG | ||||||
| STR1035-13F | AAGCTGGAATCAACCATAAACT | 55 | (AG)5 | 124 | 5’HEX | SQLE | downstream |
| STR1035-13R | AGAGCAGGAGAAACCCTAGAA | ||||||
| STR1035-14F | TGCTAAAAAGGCTGGTGATATC | 57 | (AG)11*(A)10 | 111 | 5’HEX | DXS | upstream |
| STR1035-14R | CGGCTTTCACTGTTTCACATA | ||||||
| STR1035-15F | CAAATAATATGATCCCTACAGAAGA | 56 | (TTA)4 | 191 | 5’6-FAM | HMGS | upstream |
| STR1035-15R | TAATAATAGCAGTTCCACATTCAGT | ||||||
| STR1035-18F | GCAGAAACTGTACCATGAGGAG | 57 | (CAAA)3 | 268 | 5’6-FAM | FNTA | downstream |
| STR1035-18R | CGTCTTGCTTGATTAACTAGGATT | ||||||
| STR1035-22F | CGATCCGAATCCTCTGTTAAA | 56 | (CT)5 | 191 | 5’6-FAM | MVD | upstream |
| STR1035-22R | GTCACCATTAGGATCCATTTCT | ||||||
| STR1150-1F | CAAGCTATTCGCCGTCCT | 56 | (CGC)4*(ACG)4 | 427 | 5’6-FAM | HMGR | upstream |
| STR1150-1R | CTGCCCCAGAATCGAGC | ||||||
| STR1150-3F | ATCTCCACGCCTTCCCTT | 57 | (CCA)4 | 170 | 5’6-FAM | ispD | upstream |
| STR1150-3R | CTCTGCTTCTCTTTTCGCAAT | ||||||
| STR1150-4F | AGGATAACTAACAAAAGAGAGGATG | 56 | (TC)5 | 190 | 5’6-FAM | ispE | upstream |
| STR1150-4R | TCTTCCTATAGAGGTTGAGTGCT | ||||||
| STR1150-7F | TGCCCCCCCTCATCTC | 56 | (TC)5 | 140 | 5’6-FAM | TGL4 | downstream |
| STR1150-7R | GGAAATTCTTGAGCTTGCAGT | ||||||
| STR1150-9F | GTGCCCGTTCCATTCAAG | 57 | (GA)10 | 119 | 5’6-FAM | MVK | upstream |
| STR1150-9R | TGCTCGCCGGAGAGTATG |
| Loci | Na | Ne | I | PIC | Ho | He | Nei (h) | HWE | FIS | FST | FIT | Nm |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1035P9 | 9 | 3.0711 | 1.3396 | 0.6202 | 0.5515 | 0.6769 | 0.6744 | 0.11 | −0.0311 | 0.1983 | 0.1734 | 1.0105 |
| 1035P13 | 5 | 1.5449 | 0.5833 | 0.3102 | 0.2132 | 0.354 | 0.3527 | ** | 0.3055 | 0.1281 | 0.3945 | 1.7010 |
| 1035P15 | 7 | 3.1617 | 1.3559 | 0.6505 | 0.1544 | 0.6862 | 0.6837 | ** | 0.7419 | 0.1781 | 0.7879 | 1.1533 |
| 1035P18 | 11 | 1.4961 | 0.821 | 0.3207 | 0.1103 | 0.3328 | 0.3316 | ** | 0.5952 | 0.1091 | 0.6393 | 2.0421 |
| 1035P22 | 13 | 4.9541 | 1.8423 | 0.7684 | 0.3235 | 0.8011 | 0.7981 | ** | 0.5045 | 0.1131 | 0.5605 | 1.9598 |
| 1150P1 | 12 | 2.5313 | 1.4029 | 0.5785 | 0.1838 | 0.6072 | 0.6049 | ** | 0.6646 | 0.1145 | 0.7030 | 1.9332 |
| 1150P3 | 12 | 3.3544 | 1.4452 | 0.6559 | 0.5368 | 0.7045 | 0.7019 | ** | 0.1581 | 0.0866 | 0.2310 | 2.6380 |
| 1150P4 | 5 | 2.3126 | 0.9829 | 0.4781 | 0.1691 | 0.5697 | 0.5676 | ** | 0.6610 | 0.1720 | 0.7193 | 1.2035 |
| 1150P7 | 5 | 1.1018 | 0.2441 | 0.0842 | 0.0441 | 0.0927 | 0.0924 | ** | 0.4801 | 0.0855 | 0.5246 | 2.6727 |
| 1150P9 | 20 | 4.3902 | 2.1041 | 0.7585 | 0.6912 | 0.7751 | 0.7722 | 0.02 | −0.0132 | 0.1182 | 0.1065 | 1.8653 |
| Pop | Major Allele Frequency | Genotype Number | Na | Ne | Gene Diversity | PIC | I | Ho | He | Nei (h) |
|---|---|---|---|---|---|---|---|---|---|---|
| LS | 0.58 | 4.9 | 4.4 | 2.7809 | 0.5531 | 0.5078 | 1.0689 | 0.3000 | 0.5685 | 0.5496 |
| DL | 0.61 | 4.1 | 3.2 | 2.2729 | 0.4635 | 0.4124 | 0.8135 | 0.2353 | 0.4718 | 0.4580 |
| KM | 0.66 | 5.3 | 4.7 | 2.4983 | 0.4593 | 0.4315 | 0.9585 | 0.3200 | 0.4759 | 0.4600 |
| QJL | 0.56 | 5.5 | 4.9 | 2.7692 | 0.5553 | 0.5073 | 1.0883 | 0.3733 | 0.5740 | 0.5549 |
| QJS | 0.59 | 6.0 | 5.0 | 2.4917 | 0.5489 | 0.5092 | 1.0906 | 0.3800 | 0.5678 | 0.5489 |
| ML | 0.58 | 4.3 | 3.4 | 2.3365 | 0.5087 | 0.4419 | 0.8852 | 0.3133 | 0.5262 | 0.5087 |
| JD | 0.63 | 4.9 | 4.4 | 2.2769 | 0.4801 | 0.4343 | 0.9317 | 0.2687 | 0.5016 | 0.4859 |
| TC | 0.75 | 2.3 | 2.2 | 1.6859 | 0.3278 | 0.2782 | 0.5346 | 0.2889 | 0.3471 | 0.3278 |
| YX | 0.69 | 2.6 | 2.4 | 1.9034 | 0.4094 | 0.3509 | 0.671 | 0.2375 | 0.4367 | 0.4094 |
| CX | 0.67 | 4.0 | 3.3 | 2.1356 | 0.4343 | 0.3905 | 0.7938 | 0.2182 | 0.4550 | 0.4343 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, X.; Su, Q.; Yao, B.; Yang, W.; Ma, W.; Yang, B.; Liu, C. Development of EST-SSR Markers Related to Polyphyllin Biosynthesis Reveals Genetic Diversity and Population Structure in Paris polyphylla. Diversity 2022, 14, 589. https://doi.org/10.3390/d14080589
Gao X, Su Q, Yao B, Yang W, Ma W, Yang B, Liu C. Development of EST-SSR Markers Related to Polyphyllin Biosynthesis Reveals Genetic Diversity and Population Structure in Paris polyphylla. Diversity. 2022; 14(8):589. https://doi.org/10.3390/d14080589
Chicago/Turabian StyleGao, Xiaoyang, Qixuan Su, Baolin Yao, Wenjing Yang, Weisi Ma, Bin Yang, and Changning Liu. 2022. "Development of EST-SSR Markers Related to Polyphyllin Biosynthesis Reveals Genetic Diversity and Population Structure in Paris polyphylla" Diversity 14, no. 8: 589. https://doi.org/10.3390/d14080589
APA StyleGao, X., Su, Q., Yao, B., Yang, W., Ma, W., Yang, B., & Liu, C. (2022). Development of EST-SSR Markers Related to Polyphyllin Biosynthesis Reveals Genetic Diversity and Population Structure in Paris polyphylla. Diversity, 14(8), 589. https://doi.org/10.3390/d14080589