
Genetic Diversity of Wisent Bison bonasus Based on STR Loci Analyzed in a Large Set of Samples

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
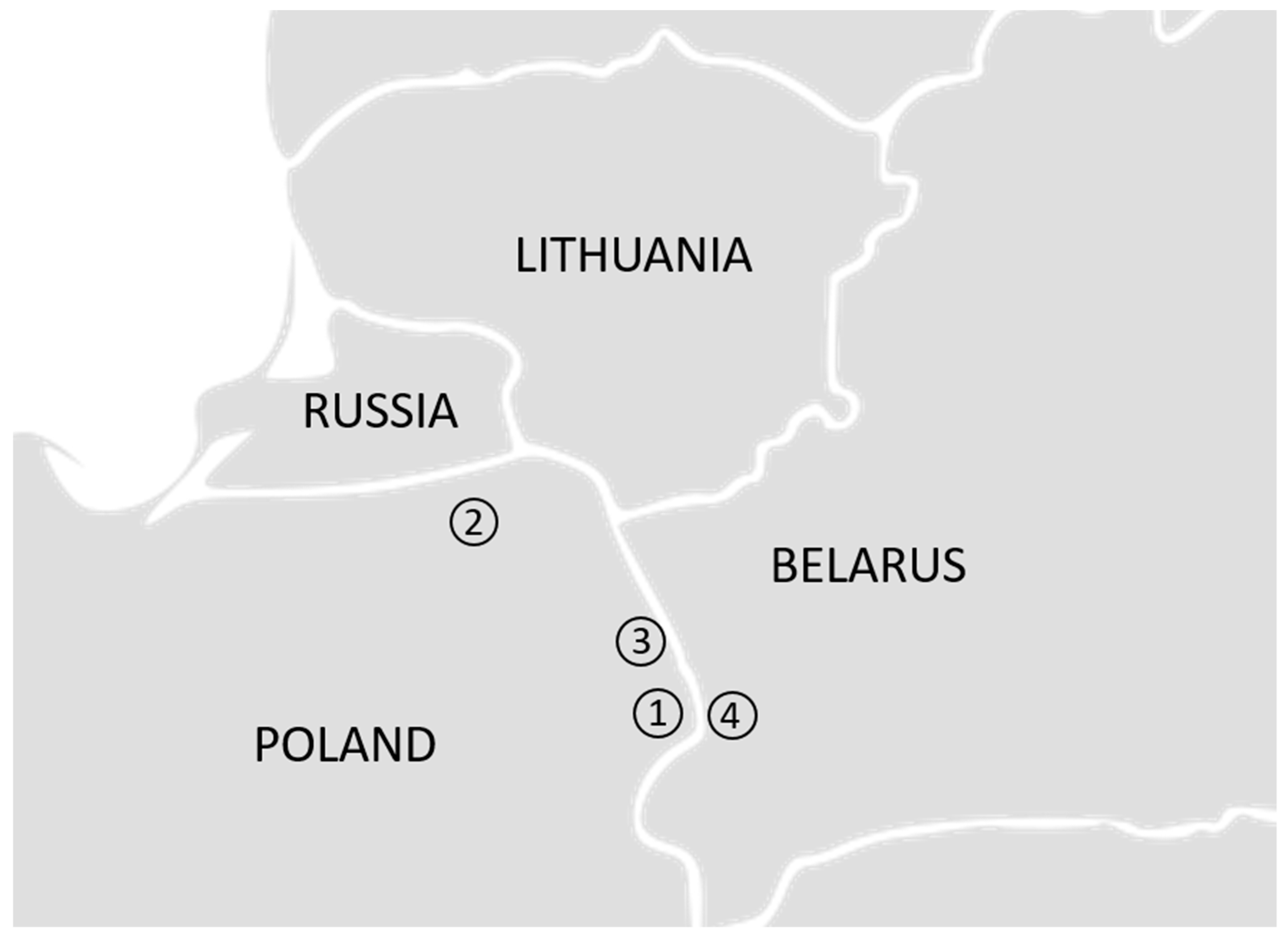
2.1. Samples and DNA Extraction
2.2. Genotyping
2.3. Statistical Analysis
3. Results
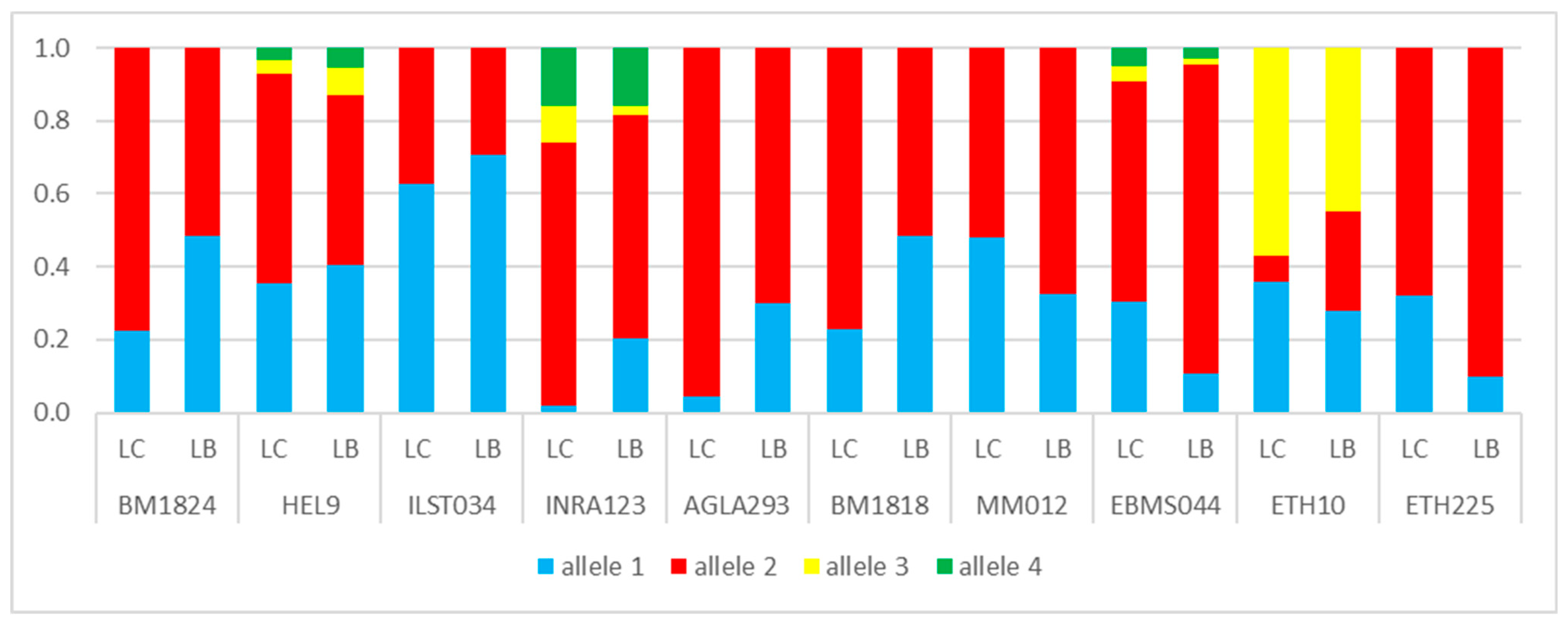
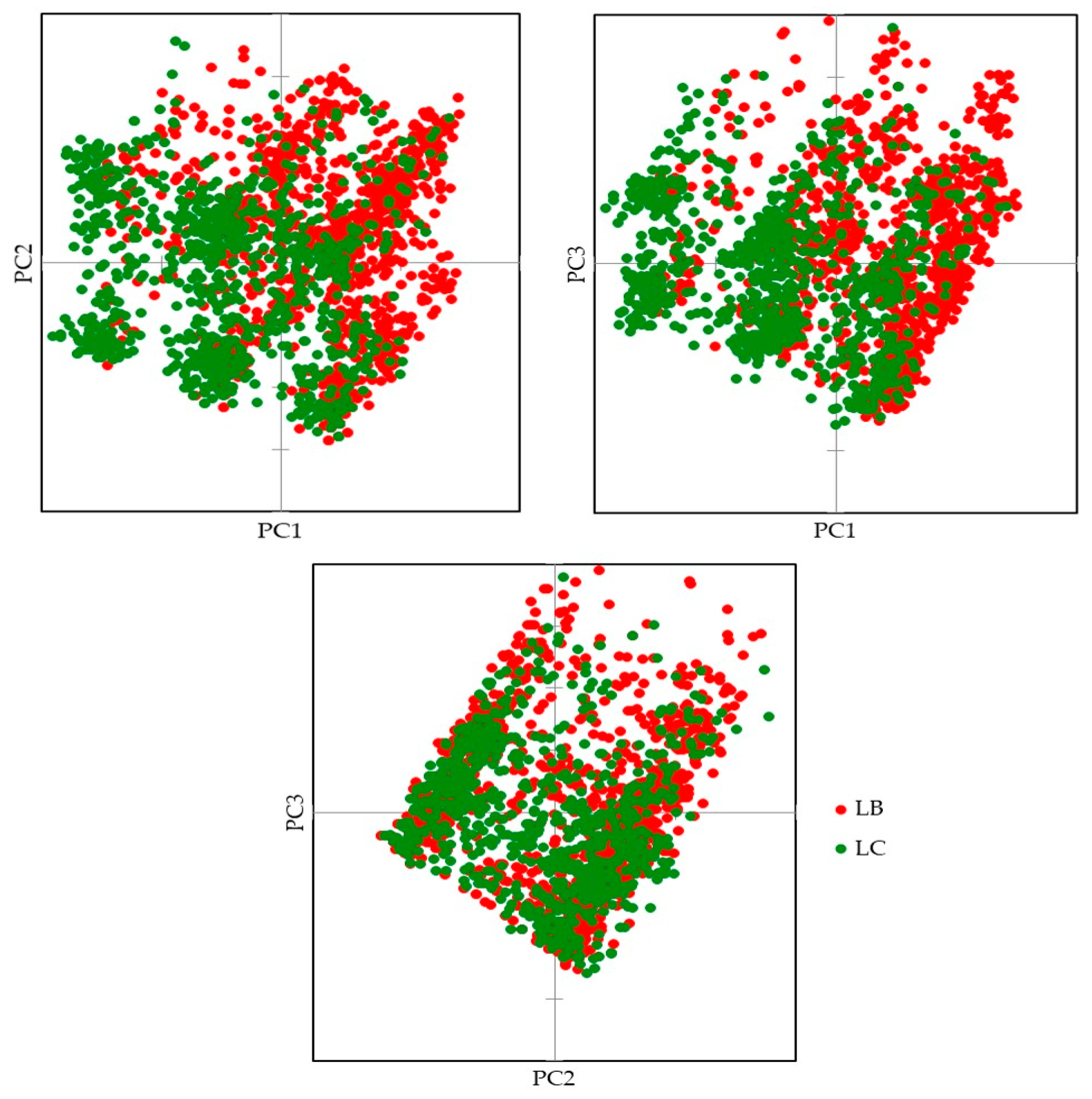
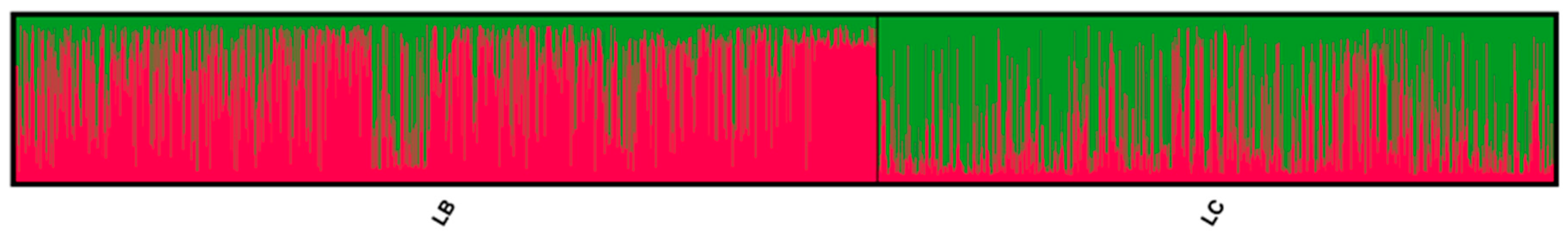
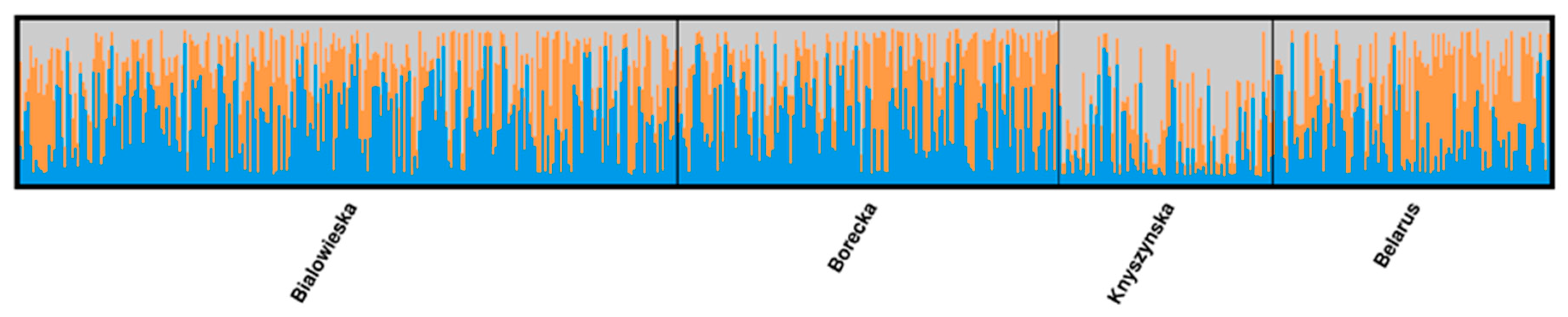
3.1. Genetic Lines—LC vs. LB
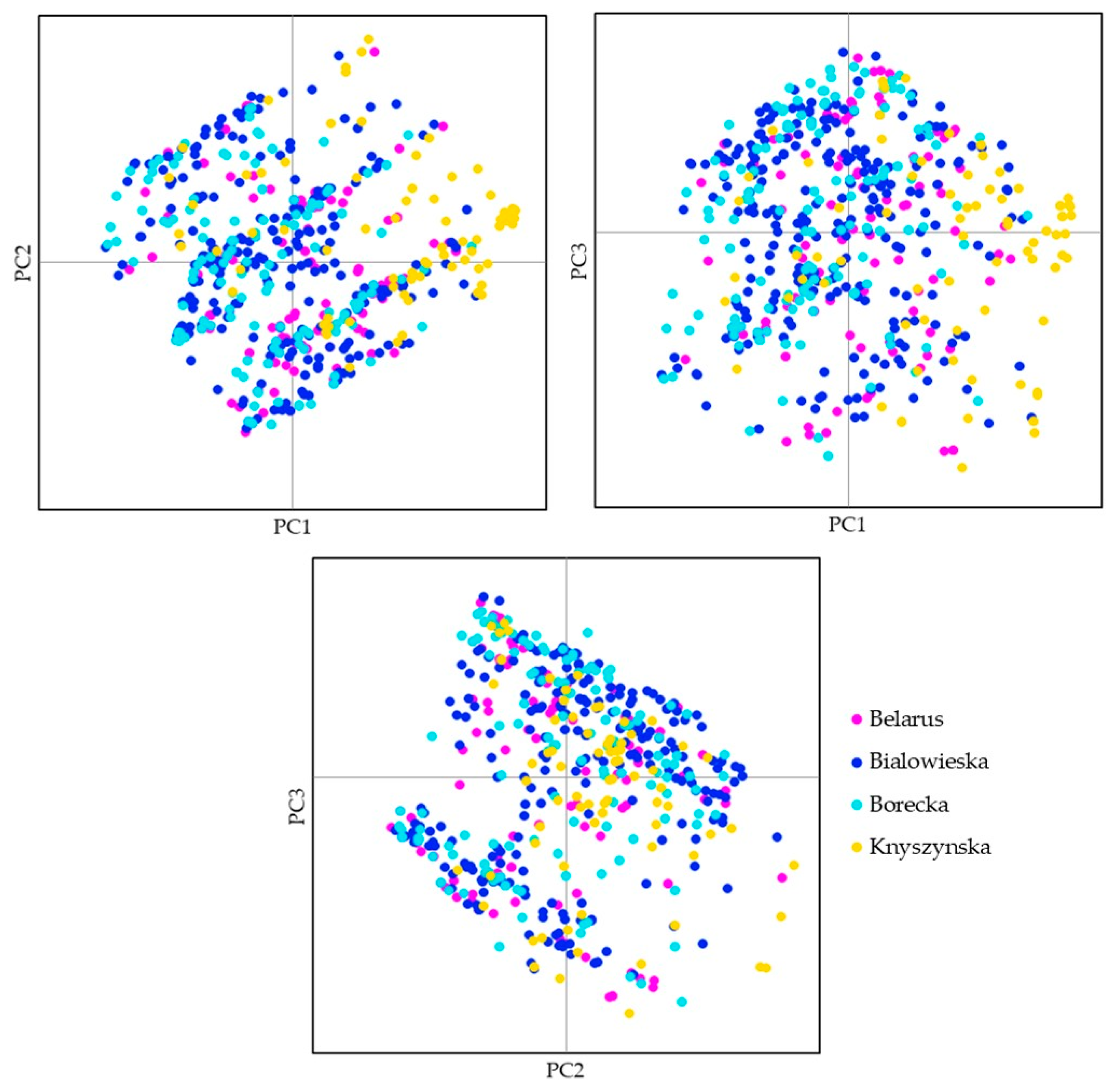
3.2. Four Populations within the Lowland Line (LB)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pucek, Z.; Belousova, I.P.; Krasińska, M.; Krasiński, Z.A.; Olech, W. European Bison Status Survey and Conservation Action Plan; IUCN The World Conservation Union: Gland, Switzerland, 2004; pp. 1–49. [Google Scholar]
- Krasińska, M.; Krasiński, Z.A. European Bison–Nature Monograph; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–379. [Google Scholar]
- Krasiński, Z.A. Free-living European bison. Acta Theriol. 1967, 12, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Krasiński, Z.A.; Krasińska, M. Free ranging European bison in Borecka Forest. Acta Theriol. 1992, 37, 301–317. [Google Scholar] [CrossRef] [Green Version]
- Krasiński, Z.A.; Krasińska, M.; Leniec, H. Żubry w Puszczy Knyszyńskiej. Park. Nar. I Rezerw. Przyr. 1994, 13, 107–114. (In Polish) [Google Scholar]
- Kozlo, P. Analysis of reintroduction and formation of European bison populations in Belarus. Eur. Bison Conserv. Newsl. 2013, 6, 21–36. [Google Scholar]
- Raczynski, J.; Bołbot, M. European Bison Pedigree Book 2021; Białowieski National Park: Białowieża, Poland, 2022; pp. 1–80. [Google Scholar]
- Olech, W.; Perzanowski, K. European Bison (Bison bonasus) Strategic Species Status Review 2020; IUCN SSC Bison Specialist Group and European Bison Conservation Center: Warsaw, Poland, 2022; pp. 1–148. [Google Scholar]
- Slatis, H.M. An analysis of inbreeding in the European bison. Genetics 1960, 45, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Olech, W. The participation of ancestral genes in the existing population of European bison. Acta Theriol. 1989, 34, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Olech, W. The number of ancestors and their contribution to European bison (Bison bonasus L.) population. Ann. Warsaw Agric. Univ. Anim. Sci. 1999, 35, 111–117. [Google Scholar]
- Olech, W. The Influence of Individual and Maternal Inbreeding on the Survival of European Bison Calves; Warsaw University of Life Sciences: Warsaw, Poland, 2003; pp. 1–78. (In Polish) [Google Scholar]
- Olech, W. The analysis of European bison genetic diversity using pedigree data. In Health Threats for the European Bison Particularly in Free-Roaming Populations in Poland; Kita, J., Anusz, K., Eds.; The SGGW Publishers: Warszawa, Poland, 2006; pp. 205–236. [Google Scholar]
- Olech, W.; Perzanowski, K. A genetic background for reintroduction program of the European bison in the Carpathians. Biol. Conserv. 2002, 108, 221–228. [Google Scholar] [CrossRef]
- Olech, W. The changes of founders’ numbers and their contribution to the European bison population during 80 years of species’ restitution. Eur. Bison Conserv. Newsl. 2009, 2, 54–60. [Google Scholar]
- Hartl, G.B.; Pucek, Z. Genetic depletion in the European bison (Bison bonasus) and the significance of electrophoretic heterozygosity for conservation. Conserv. Biol. 1994, 8, 167–174. [Google Scholar] [CrossRef]
- Gębczyński, M.; Tomaszewska-Gruszkiewicz, K. Genetic variability in the European bison. Biochem. Syst. Ecol. 1987, 15, 285–288. [Google Scholar] [CrossRef]
- Burzyńska, B.; Olech, W.; Topczewski, J. Phylogeny and genetic variation of the European bison Bison bonasus based on mitochondrial DNA D-loop sequences. Acta Theriol. 1999, 44, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Gralak, B.; Krasińska, M.; Niemczewski, C.; Krasiński, Z.A.; Żurkowski, M. Polymorphism of bovine microsatllite DNA sequences in the lowland European bison. Acta Theriol. 2004, 49, 449–456. [Google Scholar] [CrossRef]
- Luenser, K.; Fickel, J.; Lehnen, A.; Speck, S.; Ludwig, A. Low level of genetic variability in European bison (Bison bonasus) from Bialowieża National Park in Poland. Eur. J. Wildl. Res. 2005, 51, 84–87. [Google Scholar] [CrossRef]
- Roth, T.; Pfeiffer, I.; Weising, K.; Brenig, B. Application of bovine microsatellite markers for genetic diversity analysis of European bison (Bison bonasus). J. Anim. Breed. Genet. 2006, 123, 406–409. [Google Scholar] [CrossRef]
- Łopienska, M.; Nowak, Z.; Charon, K.M.; Olech, W. A comparison of polymorphism of DQA genes in European bison belonging to two genetic lines: Lowland and Lowland-Caucasian. Ann. Wars. Univ. Life Sci. SGGW Anim. Sci. 2011, 49, 93–102. [Google Scholar]
- Tokarska, M.; Bunevich, A.N.; Demontis, D.; Sipko, T.; Perzanowski, K.; Baryshnikov, G.; Kowalczyk, R.; Voitukhovskaya, Y.; Wójcik, J.M.; Marczuk, B.; et al. Genes of the extinct Caucasian bison still roam the Białowieża Forest and are the source of genetic discrepances between Polish and Belarusian populations of the European bison. Bison bonasus. Biol. J. Linn. Soc. 2015, 114, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Dotsev, A.V.; Aksenova, P.V.; Volkova, V.V.; Kharzinova, V.R.; Kostyunina, O.V.; Mnatsekanov, R.A.; Zinovieva, N.A. Study of Allele Pool and Genetic Structure of Russian Population of Lowland-Caucasian Line of European Bison (Bison bonasus). Russ. J. Genet. Appl. Res. 2018, 8, 31–36. [Google Scholar] [CrossRef]
- Homel, K.V.; Sliwinska, A.A.; Valnisty, A.A.; Nikifirov, M.E. New data on the genetic diversity of European bison (Bison bonasus) in Belarus. Theriol. Ukr. 2020, 19, 45–53. [Google Scholar] [CrossRef]
- Kostyunina, O.V.; Mikhailova, A.D.; Dotseva, A.V.; Zemlyanko, I.I.; Volkova, V.V.; Fornara, M.S.; Akopyana, N.A.; Kramarenko, A.S.; Okhlopkove, I.M.; Aksenova, P.V.; et al. Comparative Genetic Characteristics of the Russian and Belarusian Populations of Wisent (Bison bonasus), North American Bison (Bison bison) and Cattle (Bos taurus). Cytol. Genet. 2020, 54, 116–123. [Google Scholar] [CrossRef]
- Nguye, T.T.; Genini, S.; Menetrey, F.; Malek, M.; Vogeli, P.; Goe, M.R.; Stranzinger, G. Application of bovine microsatellite markers for genetic diversity analysis of Swiss yak (Poephagus grunniens). Anim. Genet. 2005, 36, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Mukherjee, A.; Kumar, S.; Verma, H.; Bhardwaj, S.; Togla, O.; Joardar, S.N.; Longkumer, I.; Mech, M.; Khate, K.; et al. Genetic Characterization of Endangered Indian Mithun (Bos frontalis), Indian Bison/Wild Gaur (Bos gaurus) and Tho-tho Cattle (Bos indicus) Populations Using SSR Markers Reveals Their Diversity and Unique Phylogenetic Status. Diversity 2022, 14, 548. [Google Scholar] [CrossRef]
- Crestanello, B.; Pechioli, E.; Vernesi, C.; Mona, S.; Martinkova, N.; Janiga, M.; Hauffe, H.C.; Bertorelle, G. The genetic impact of translocations and habitat fragmentation in chamois (Rupicapra) spp. J. Hered. 2009, 100, 691–708. [Google Scholar] [CrossRef] [Green Version]
- Mommens, G.; Peelman, L.J.; Van Zeveren, A.; D’Ieteren, G.; Wissocq, N. Microsatellite variation between an African and five European taurine breeds results in a geographical phylogenetic tree with a bison outgroup. J. Anim. Breed. Genet. 1999, 116, 325–330. [Google Scholar] [CrossRef]
- Mommens, G.; Zeveren, A.; Peelman, L.J. Effectiveness of bovine microsatellites in resolving paternity cases in American bison, Bison bison L. Anim. Genet. 1998, 29, 12–18. [Google Scholar] [CrossRef]
- Schnabel, R.D.; Ward, T.J.; Derr, J.N. Validation of 15 microsatellites for parentage testing in North American bison, Bison bison and domestic cattle. Anim. Genet. 2000, 31, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Halbert, N.; Gogan, P.J.P.; Hedrick, P.W.; Wahl, J.M.; Derr, J.N. Genetic Population Substructure in Bison at Yellowstone National Park. J. Hered. 2012, 103, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Halbert, N.D.; Derr, J.N. A Comprehensive Evaluation of Cattle Introgression into US Federal Bison Herds. J. Hered. 2007, 98, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ranglack, D.H.; Dobson, L.R.; Du Toit, J.H.; Derr, J. Genetic Analysis of the Henry Mountains Bison Herd. PLoS ONE 2015, 10, e0144239. [Google Scholar] [CrossRef] [Green Version]
- Tokarska, M.; Marshall, T.; Kowalczyk, R.; Wójcik, J.M.; Pertoldi, C.; Kristensen, T.N.; Loeschecke, V.; Gregersen, V.R.; Bendixen, C. Effectiveness of microsatellite and SNP markers for parentage and identity analysis in species with low genetic diversity: The case of European bison. Heredity 2009, 103, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Kamiński, S.; Olech, W.; Oleński, K.; Nowak, Z.; Ruść, A. Single nucleotide polymorphisms between two lines of European bison (Bison bonasus) detected by the use of Illumina Bovine 50 K BeadChip. Conserv. Genet. Resour. 2012, 4, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Wojciechowska, M.; Nowak, Z.; Gurgul, A.; Olech, W.; Drobik, W.; Szmatoła, T. Panel of informative SNP markers for two genetic lines of European bison: Lowland and Lowland–Caucasian. Anim. Biodivers. Conserv. 2017, 40, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Wojciechowska, M.; Puchała, K.; Nowak-Życzyńska, Z.; Perlińska-Teresiak, M.; Kloch, M.; Drobik-Czwarno, W.; Olech, W. From Wisent to the Lab and Back Again—A Complex SNP Set for Population Management as an Effective Tool in European Bison Conservation. Diversity 2023, 15, 116. [Google Scholar] [CrossRef]
- Oleński, K.; Kamiński, S.; Tokarska, M.; Hering, D.M. Subset of SNPs for parental identification in European bison Lowland-Białowieża line (Bison bonasus bonasus). Conserv. Genet. Resour. 2018, 10, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Grzegrzółka, B.; Olech, W.; Krasiński, Z.A. Struktura genetyczna wolnych stad żubrów nizinnych w Polsce. Park. Nar. I Rezerw. Przyr. 2004, 23, 665–677. (In Polish) [Google Scholar]
- Krasińska, M.; Krasiński, Z.A.; Bunevich, A.N. Spatial distribution of the European bison population in Polish and Belarussian parts of Białowieża Forest. In Proceedings of the Conference European Bison Conservation, Bialowieza, Poland, 30 September–2 October 2004; p. 73. [Google Scholar]
- Barendse, W.; Armitage, S.M.; Kossarek, L.M.; Shalom, A.; Kirkpatrick, B.W.; Ryan, A.M.; Clayton, D.; Li, L.; Neibergs, H.L.; Zhang, N.; et al. A genetic linkage map of the bovine genome. Nat. Genet. 1994, 6, 227–235. [Google Scholar] [CrossRef]
- Kaukinen, J.; Varvio, S.L. Eight polymorphic bovine microsatellites. Anim. Genet. 1993, 24, 148. [Google Scholar] [CrossRef]
- FAO. Global Project for the Maintenance of Domestic Animal Genetic Diversity (MoDAD)–Draft Project Formulation Report; FAO: Rome, Italy, 1995. [Google Scholar]
- Vaiman, D.; Mercier, D.; Moazami-Goudarzi, K.; Eggen, A.; Ciampolini, R.; Lépingle, A.; Velmala, R.; Kaukinen, J.; Varvio, S.L.; Martin, P.; et al. A set of 99 cattle microsatellites: Characterization, synteny mapping, and polymorphism. Mamm. Genome 1994, 5, 288–297. [Google Scholar] [CrossRef]
- Mommens, G.; Coppieters, W.; Van de Weghe, A.; Van Zeveren, A.; Bouquet, Y. Dinucleotide repeat polymorphism at the bovine MM12E6 and MM8D3 loci. Anim. Genet. 1994, 25, 368. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.D.; Kappes, S.M.; Keele, J.W.; Stone, R.T.; Sunden, S.L.; Hawkins, G.A.; Toldo, S.S.; Fries, R.; Grosz, M.D.; Yoo, J.; et al. A genetic linkage map for cattle. Genetics 1994, 136, 619–639. [Google Scholar] [CrossRef]
- Toldo, S.S.; Fries, R.; Steffen, P.; Neibergs, H.L.; Barendse, W.; Womack, J.E.; Hetzel, D.J.; Stranzinger, G. Physically mapped, cosmid-derived microsatellite markers as anchor loci on bovine chromosomes. Mamm. Genome 1993, 4, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Steffen, P.; Eggen, A.; Dietz, A.B.; Womack, J.E.; Stranzinger, G.; Fries, R. Isolation and mapping of polymorphic microsatellites in cattle. Anim. Genet. 1993, 24, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research_an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team 2022, RStudio: Integrated Development Environment for R. RStudio, PBC, Boston, MA URL. Available online: http://www.rstudio.com/ (accessed on 2 March 2022).
- Ligges, U.; Mächler, M. Scatterplot3d–an R Package for Visualizing Multivariate Data. J. Stat. Softw. 2003, 8, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Gen. Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Eco. Res. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Cronin, M.A.; MacNeil, M.D.; Leeburg, V.; Vu, N.; Blackburn, H.D.; Derr, J. Genetic variation and differentiation of bison (Bison bison)) subspecies’ and cattle (Bos taurus) breeds and subspecies. J. Hered. 2013, 104, 500–509. [Google Scholar] [CrossRef] [Green Version]
- Wilson, G.A.; Strobeck, C. The isolation and characterization of microsatellite loci in bison, and their usefulness in other artiodactyls. Anim. Genet. 1999, 30, 226–227. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Samples | Number of Samples | Genetic Line * | Number of Loci | Allelic Richness | Observed Heterozygosity | Expected Heterozygosity | Authors |
|---|---|---|---|---|---|---|---|
| Poland Białowieska | 22 | LB | 21 | 1.905 | 0.241 | 0.245 | Gralak et al., 2004 [19] |
| Poland Białowieska | 38 | LB | 9 | 2.667 | 0.506 | 0.507 | Luenser et al., 2005 [20] |
| Poland Białowieska | 247 | LB | 17 | 2.790 | 0.259 | 0.279 | Tokarska et al., 2015 [23] |
| Belarus | 48 | LB | 17 | 2.470 | 0.283 | 0.270 | Tokarska et al., 2015 [23] |
| Belarus | 30 | LB | 11 | 2.434 | 0.424 | 0.317 | Homel et al., 2020 [25] |
| Belarus | 42 | LB | 11 | 1.818 | 0.281 | 0.277 | Kostyunina et al., 2020 [26] |
| Russia reserves | 111 | LC | 11 | 2.182 | 0.302 | 0.286 | Kostyunina et al., 2020 [26] |
| Poland Bieszczady and Russia | 56 | LC | 17 | 3.260 | 0.358 | 0.392 | Tokarska et al., 2015 [23] |
| Russian reserves | 26 | LC | 10 | 2.800 | 0.262 | 0.293 | Dotsev et al., 2018 [24] |
| German reserves | 35 | LC | 11 | 2.360 | 0.453 | 0.447 | Roth et al., 2006 [21] |
| Locus | Forward Primer Sequence (5′–3′) | Reverse Primer Sequence (5′–3′) | Fluorescent Dye | Panel | Source |
|---|---|---|---|---|---|
| BM1824 | GAGCAAGGTGTTTTTCCAATC | CATTCTCCAACTGCTTCCTTG | PET | 55 | [43] |
| HEL009 | CCCATTCAGTCTTCAGAGGT | CACATCCATGTTCTCACCAC | VIC | 55 | [44] |
| ILST034 | AAGGGTCTAATGCCACTGGC | GACCTGGTTTAGCAGAGAGC | 6-FAM | 55 | [45] |
| INRA123 | TCTAGAGGATCCCCGCTGAC | AGAGAGCAACTCCACTGTGC | NED | 55 | [46] |
| MM012 | CAAGACAGGTGTTTCAATCT | ATCGACTCTGGGGATGATGT | 6-FAM | 55 | [47] |
| AGLA293 | GAAACTCAACCCAAGACAACTCAAG | ATGACTTTATTCTCCACCTAGCAGA | VIC | 55 | [31] |
| BM1818 | AGCTGGGAATATAACCAAAGG | AGTGCTTTCAAGGTCCATGC | 6-FAM | 55 | [48] |
| EBMS044 | CCTTGCCACTATTTCCTCCA | CCAAATGACACATGACAGCC | NED | 60 | [45] |
| ETH010 | GTTCAGGACTGGCCCTGCTAACA | CCTCCAGCCCACTTTCTCTTCTC | NED | 60 | [49] |
| ETH225 | GATCACCTTGCCACTATTTCCT | ACATGACAGCCAGCTGCTACT | VIC | 60 | [50] |
| LC Line | LB Line | ||||||
|---|---|---|---|---|---|---|---|
| Locus | Na | Ne | Ho | He | Ne | Ho | He |
| BM1824 | 2 | 1.998 | 0.440 | 0.500 | 1.538 | 0.294 | 0.350 |
| HEL009 | 4 | 2.568 | 0.241 | 0.611 | 2.189 | 0.173 | 0.543 |
| ILST034 | 2 | 1.706 | 0.383 | 0.414 | 1.882 | 0.394 | 0.469 |
| INRA123 | 4 | 2.257 | 0.601 | 0.557 | 1.786 | 0.521 | 0.440 |
| AGLA293 | 2 | 1.719 | 0.389 | 0.418 | 1.090 | 0.080 | 0.083 |
| BM1818 | 2 | 1.998 | 0.444 | 0.499 | 1.546 | 0.304 | 0.353 |
| MM012 | 2 | 1.783 | 0.402 | 0.439 | 1.997 | 0.421 | 0.499 |
| EBMS044 | 4 | 1.377 | 0.204 | 0.274 | 2.168 | 0.452 | 0.539 |
| ETH010 | 3 | 2.833 | 0.576 | 0.647 | 2.185 | 0.476 | 0.542 |
| ETH225 | 2 | 1.217 | 0.122 | 0.178 | 1.771 | 0.364 | 0.435 |
| Mean | 2.7 | 1.946 | 0.380 | 0.454 | 1.815 | 0.348 | 0.425 |
| SE | 0.3 | 0.159 | 0.049 | 0.046 | 0.111 | 0.044 | 0.044 |
| Białowieska | Borecka | Knyszyńska | Belarus | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ne | Ho | He | Ne | Ho | He | Ne | Ho | He | Ne | Ho | He | |
| BM1824 | 1.63 | 0.36 | 0.39 | 1.37 | 0.27 | 0.27 | 1.84 | 0.29 | 0.46 | 1.75 | 0.43 | 0.43 |
| HEL009 | 2.17 | 0.18 | 0.54 | 2.34 | 0.23 | 0.57 | 2.14 | 0.34 | 0.53 | 2.52 | 0.23 | 0.60 |
| ILST034 | 1.63 | 0.35 | 0.38 | 1.29 | 0.21 | 0.22 | 1.54 | 0.35 | 0.35 | 1.95 | 0.42 | 0.49 |
| INRA123 | 1.87 | 0.58 | 0.46 | 1.72 | 0.51 | 0.42 | 1.87 | 0.58 | 0.47 | 1.53 | 0.39 | 0.34 |
| AGLA293 | 1.15 | 0.14 | 0.13 | 1.00 | 0 | 0 | 1.01 | 0.01 | 0.01 | 1.19 | 0.13 | 0.16 |
| BM1818 | 1.67 | 0.40 | 0.40 | 1.43 | 0.26 | 0.30 | 1.88 | 0.33 | 0.47 | 1.76 | 0.44 | 0.43 |
| MM012 | 1.85 | 0.42 | 0.46 | 1.93 | 0.41 | 0.48 | 1.19 | 0.10 | 0.16 | 1.88 | 0.48 | 0.47 |
| EBMS044 | 2.42 | 0.50 | 0.59 | 2.20 | 0.49 | 0.55 | 1.74 | 0.19 | 0.43 | 2.44 | 0.63 | 0.59 |
| ETH010 | 2.09 | 0.53 | 0.52 | 2.09 | 0.52 | 0.52 | 1.29 | 0.19 | 0.22 | 2.33 | 0.54 | 0.57 |
| ETH225 | 1.96 | 0.47 | 0.49 | 1.96 | 0.46 | 0.49 | 1.45 | 0.12 | 0.31 | 1.76 | 0.34 | 0.43 |
| Mean | 1.84 | 0.39 | 0.44 | 1.73 | 0.33 | 0.38 | 1.60 | 0.25 | 0.34 | 1.91 | 0.40 | 0.45 |
| s.d. | 0.11 | 0.05 | 0.04 | 0.14 | 0.05 | 0.06 | 0.11 | 0.05 | 0.05 | 0.13 | 0.05 | 0.04 |
| Belarus | Białowieska | Borecka | Knyszyńska | |
|---|---|---|---|---|
| Belarus | - | 0.80% | 2.61% | 7.81% |
| Białowieska | 1.39% | - | 1.11% | 8.21% |
| Borecka | 3.14% | 0.92% | - | 9.85% |
| Knyszyńska | 12.06% | 12.78% | 15.19% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olech, W.; Wojciechowska, M.; Kloch, M.; Perlińska-Teresiak, M.; Nowak-Życzyńska, Z. Genetic Diversity of Wisent Bison bonasus Based on STR Loci Analyzed in a Large Set of Samples. Diversity 2023, 15, 399. https://doi.org/10.3390/d15030399
Olech W, Wojciechowska M, Kloch M, Perlińska-Teresiak M, Nowak-Życzyńska Z. Genetic Diversity of Wisent Bison bonasus Based on STR Loci Analyzed in a Large Set of Samples. Diversity. 2023; 15(3):399. https://doi.org/10.3390/d15030399
Chicago/Turabian StyleOlech, Wanda, Marlena Wojciechowska, Marta Kloch, Magdalena Perlińska-Teresiak, and Zuza Nowak-Życzyńska. 2023. "Genetic Diversity of Wisent Bison bonasus Based on STR Loci Analyzed in a Large Set of Samples" Diversity 15, no. 3: 399. https://doi.org/10.3390/d15030399
APA StyleOlech, W., Wojciechowska, M., Kloch, M., Perlińska-Teresiak, M., & Nowak-Życzyńska, Z. (2023). Genetic Diversity of Wisent Bison bonasus Based on STR Loci Analyzed in a Large Set of Samples. Diversity, 15(3), 399. https://doi.org/10.3390/d15030399