
Whole Genome Resequencing Reveals Genetic Diversity and Selection Signatures of Ethiopian Indigenous Cattle Adapted to Local Environments

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cattle Populations and Sample Collection
2.2. Whole Genome Sequences, Reads Mapping, and SNPs Calling
2.3. Population Genetic Structure
2.4. Genomic Diversity
2.4.1. Nucleotide Diversity, Population Genetic Differentiation, and Heterozygosity
2.4.2. Runs of Homozygosity-Based Genomic Inbreeding Coefficient (Froh)
2.4.3. Linkage Disequilibrium (LD) and Effective Population Size
2.4.4. Phylogenetic Relationships among Cattle Samples
2.5. Detection of Selection Signatures and Their Functional Annotations
3. Results
3.1. Population Genetic Structure in Ethiopian Cattle
3.2. Genomic Diversity in Ethiopian Cattle Populations
3.2.1. Nucleotide Diversity, Heterozygosity, and Population Genetic Differentiation
3.2.2. ROHs-Based Genomic Inbreeding Coefficient (Froh)
3.2.3. Linkage Disequilibrium (LD) and Effective Population Size (Ne)
3.2.4. Phylogenetic Relationships between Ethiopian and Non-Ethiopian Cattle
3.3. Selection Signatures in Ethiopian Cattle
3.3.1. Comparative Analysis of Selection Sweeps in Ethiopian Cattle
3.3.2. Annotation of Candidate Selection Signatures
4. Discussion
4.1. Selection Signatures for Immune Response
4.2. Selection Signatures for Growth and Production
4.3. Selection Signatures for Reproduction
4.4. Selection Signatures for Environmental Adaptation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edwards, C.J.; Magee, D.A.; Park, S.D.E.; McGettigan, P.A.; Lohan, A.J.; Murphy, A.; Finlay, E.K.; Shapiro, B.; Chamberlain, A.T.; Richards, M.B.; et al. A Complete Mitochondrial Genome Sequence from a Mesolithic Wild Aurochs (Bos Primigenius). PLoS ONE 2010, 5, e9255. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.Y.W.; Larson, G.; Edwards, C.J.; Heupink, T.H.; Lakin, K.E.; Holland, P.W.H.; Shapiro, B. Correlating Bayesian Date Estimates with Climatic Events and Domestication Using a Bovine Case Study. Biol. Lett. 2008, 4, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Ajmone-Marsan, P.; Garcia, J.F.; Lenstra, J.A. On the Origin of Cattle: How Aurochs Became Cattle and Colonized the World. Evol. Anthropol. 2010, 19, 148–157. [Google Scholar] [CrossRef]
- Pitt, D.; Sevane, N.; Nicolazzi, E.L.; MacHugh, D.E.; Park, S.D.E.; Colli, L.; Martinez, R.; Bruford, M.W.; Orozco-terWengel, P. Domestication of Cattle: Two or Three Events? Evol. Appl. 2019, 12, 123–136. [Google Scholar] [CrossRef]
- Chen, S.; Lin, B.Z.; Baig, M.; Mitra, B.; Lopes, R.J.; Santos, A.M.; Magee, D.A.; Azevedo, M.; Tarroso, P.; Sasazaki, S.; et al. Zebu Cattle Are an Exclusive Legacy of the South Asia Neolithic. Mol. Biol. Evol. 2010, 27, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Gifford-Gonzalez, D.; Hanotte, O. Domesticating Animals in Africa: Implications of Genetic and Archaeological Findings. J. World Prehistory 2011, 24, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Stock, F.; Gifford-Gonzalez, D. Genetics and African Cattle Domestication. Afr. Archaeol. Rev. 2013, 30, 51–72. [Google Scholar] [CrossRef]
- Hanotte, O.; Bradley, D.G.; Ochieng, J.W.; Verjee, Y.; Hill, E.W.; Rege, J.E.O. African Pastoralism: Genetic Imprints of Origins and Migrations. Science 2002, 296, 336–339. [Google Scholar] [CrossRef] [Green Version]
- Hanotte, O.; Tawah, C.L.; Bradley, D.G.; Okomo, M.; Verjee, Y.; Ochieng, J.; Rege, J.E.O. Geographic Distribution and Frequency of a Taurine Bos Taurus and an Indicine Bos Indicus Y Specific Allele amongst Sub-Saharan African Cattle Breeds. Mol. Ecol. 2000, 9, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mwai, O.; Hanotte, O.; Kwon, Y.-J.; Cho, S. African Indigenous Cattle: Unique Genetic Resources in a Rapidly Changing World. Asian-Australas. J. Anim. Sci. 2015, 28, 911–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rege, J.E.O.; Tawah, C.L. The State of African Cattle Genetic Resources II. Geographical Distribution, Characteristics and Uses of Present-Day Breeds and Strains. Anim. Genet. Resour. Inf. 1999, 26, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Rege, J.E.O. The State of African Cattle Genetic Resources I. Classification Framework and Identification of Threatened and Extinct Breeds. Anim. Genet. Resour. Inf. 1999, 25, 1–25. [Google Scholar] [CrossRef]
- Gebrehiwot, N.Z.; Strucken, E.M.; Aliloo, H.; Marshall, K.; Gibson, J.P. The Patterns of Admixture, Divergence, and Ancestry of African Cattle Populations Determined from Genome-Wide SNP Data. BMC Genom. 2020, 21, 869. [Google Scholar] [CrossRef]
- Shava, S.; Masuku, S. Living Currency: The Multiple Roles of Livestock in Livelihood Sustenance and Exchange in the Context of Rural Indigenous Communities in Southern Africa. S. Afr. J. Environ. Educ. 2019, 35, 1–13. [Google Scholar] [CrossRef]
- Barendse, W. Climate Adaptation of Tropical Cattle. Annu. Rev. Anim. Biosci. 2017, 5, 133–150. [Google Scholar] [CrossRef]
- Decker, J.E.; McKay, S.D.; Rolf, M.M.; Kim, J.; Molina Alcalá, A.; Sonstegard, T.S.; Hanotte, O.; Götherström, A.; Seabury, C.M.; Praharani, L.; et al. Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle. PLoS Genet. 2014, 10, e1004254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Kwon, T.; Dessie, T.; Yoo, D.A.; Mwai, O.A.; Jang, J.; Sung, S.; Lee, S.B.; Salim, B.; Jung, J.; et al. The Mosaic Genome of Indigenous African Cattle as a Unique Genetic Resource for African Pastoralism. Nat. Genet. 2020, 52, 1099–1110. [Google Scholar] [CrossRef]
- Whitlock, R. Relationships between Adaptive and Neutral Genetic Diversity and Ecological Structure and Functioning: A Meta-Analysis. J. Ecol. 2014, 102, 857–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadi, H.; Tibbo, M.; Takahashi, Y.; Nomura, K.; Hanada, H.; Amano, T. Microsatellite Analysis Reveals High Genetic Diversity but Low Genetic Structure in Ethiopian Indigenous Cattle Populations. Anim. Genet. 2008, 39, 425–431. [Google Scholar] [CrossRef]
- Zerabruk, M.; Li, M.H.; Kantanen, J.; Olsaker, I.; Ibeagha-Awemu, E.M.; Erhardt, G.; Vangen, O. Genetic Diversity and Admixture of Indigenous Cattle from North Ethiopia: Implications of Historical Introgressions in the Gateway Region to Africa. Anim. Genet. 2012, 43, 257–266. [Google Scholar] [CrossRef]
- Tarekegn, G.M.; Ji, X.Y.; Bai, X.; Liu, B.; Zhang, W.; Birungi, J.; Djikeng, A.; Tesfaye, K. Variations in Mitochondrial Cytochrome b Region among Ethiopian Indigenous Cattle Populations Assert Bos Taurus Maternal Origin and Historical Dynamics. Asian-Australas. J. Anim. Sci. 2018, 31, 1393. [Google Scholar] [CrossRef] [Green Version]
- Edea, Z.; Dadi, H.; Kim, S.-W.; Dessie, T.; Lee, T.; Kim, H.; Kim, J.-J.; Kim, K.-S. Genetic Diversity, Population Structure and Relationships in Indigenous Cattle Populations of Ethiopia and Korean Hanwoo Breeds Using SNP Markers. Front. Genet. 2013, 4, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roetzer, A.; Diel, R.; Kohl, T.A.; Rückert, C.; Nübel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; Rüsch-Gerdes, S.; et al. Whole Genome Sequencing versus Traditional Genotyping for Investigation of a Mycobacterium Tuberculosis Outbreak: A Longitudinal Molecular Epidemiological Study. PLoS Med. 2013, 10, e1001387. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP Calling from Next-Generation Sequencing Data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terefe, E.; Belay, G.; Han, J.; Hanotte, O.; Tijjani, A. Genomic Adaptation of Ethiopian Indigenous Cattle to High Altitude. Front. Genet. 2022, 13, 234. [Google Scholar] [CrossRef] [PubMed]
- Shamimuzzaman, M.; Le Tourneau, J.J.; Unni, D.R.; Diesh, C.M.; Triant, D.A.; Walsh, A.T.; Tayal, A.; Conant, G.C.; Hagen, D.E.; Elsik, C.G. Bovine Genome Database: New Annotation Tools for a New Reference Genome. Nucleic Acids Res. 2020, 48, D676–D681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358. [Google Scholar] [CrossRef] [PubMed]
- Kardos, M.; Luikart, G.; Allendorf, F.W. Measuring Individual Inbreeding in the Age of Genomics: Marker-Based Measures Are Better than Pedigrees. Heredity 2015, 115, 63–72. [Google Scholar] [CrossRef]
- Makgahlela, M.L.; Mäntysaari, E.A.; Strandén, I.; Koivula, M.; Nielsen, U.S.; Sillanpää, M.J.; Juga, J. Across Breed Multi-Trait Random Regression Genomic Predictions in the Nordic Red Dairy Cattle. J. Anim. Breed. Genet. 2013, 130, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; MacCiotta, N.P.P. Analysis of Runs of Homozygosity and Their Relationship with Inbreeding in Five Cattle Breeds Farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Dong, S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A Tool to Estimate Trends in Recent Effective Population Size Trajectories Using Genome-Wide SNP Data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Sved, J.A. Linkage Disequilibrium and Homozygosity of Chromosome Segments in Finite Populations. Theor. Popul. Biol. 1971, 2, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for Inferring Very Large Phylogenies by Using the Neighbor-Joining Method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, S.R.; Browning, B.L. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies by Use of Localized Haplotype Clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [Green Version]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A Map of Recent Positive Selection in the Human Genome. PLoS Biol. 2006, 4, 0446–0458. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Patterson, N.; Reich, D. Population Differentiation as a Test for Selective Sweeps. Genome Res. 2010, 20, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Xu, L.; Bickhart, D.M.; Cole, J.B.; Schroeder, S.G.; Song, J.; Van Tassell, C.P.; Sonstegard, T.S.; Liu, G.E. Genomic Signatures Reveal New Evidences for Selection of Important Traits in Domestic Cattle. Mol. Biol. Evol. 2015, 32, 711–725. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of Selection Signatures in Dairy and Beef Cattle Using High-Density Genomic Information. Genet. Sel. Evol. 2015, 47, 49. [Google Scholar] [CrossRef] [Green Version]
- Taye, M.; Kim, J.; Yoon, S.H.; Lee, W.; Hanotte, O.; Dessie, T.; Kemp, S.; Mwai, O.A.; Caetano-Anolles, K.; Cho, S.; et al. Whole Genome Scan Reveals the Genetic Signature of African Ankole Cattle Breed and Potential for Higher Quality Beef. BMC Genet. 2017, 18, 11. [Google Scholar] [CrossRef]
- Tillgren, V.; Ho, J.C.S.; Önnerfjord, P.; Kalamajski, S. The Novel Small Leucine-Rich Protein Chondroadherin-like (CHADL) Is Expressed in Cartilage and Modulates Chondrocyte Differentiation. J. Biol. Chem. 2015, 290, 918–925. [Google Scholar] [CrossRef] [Green Version]
- Sadkowski, T.; Jank, M.; Zwierzchowski, L.; Siadkowska, E.; Oprza̧dek, J.; Motyl, T. Gene Expression Profiling in Skeletal Muscle of Holstein-Friesian Bulls with Single-Nucleotide Polymorphism in the Myostatin Gene 5′-Flanking Region. J. Appl. Genet. 2008, 49, 237–250. [Google Scholar] [CrossRef]
- Hu, Y.; Xia, H.; Li, M.; Xu, C.; Ye, X.; Su, R.; Zhang, M.; Nash, O.; Sonstegard, T.S.; Yang, L.; et al. Comparative Analyses of Copy Number Variations between Bos Taurus and Bos Indicus. BMC Genom. 2020, 21, 682. [Google Scholar] [CrossRef]
- Kask, M.; Pruunsild, P.; Timmusk, T. Bidirectional Transcription from Human LRRTM2/CTNNA1 and LRRTM1/CTNNA2 Gene Loci Leads to Expression of N-Terminally Truncated CTNNA1 and CTNNA2 Isoforms. Biochem. Biophys. Res. Commun. 2011, 411, 56–61. [Google Scholar] [CrossRef]
- Lv, F.H.; Cao, Y.H.; Liu, G.J.; Luo, L.Y.; Lu, R.; Liu, M.J.; Li, W.R.; Zhou, P.; Wang, X.H.; Shen, M.; et al. Whole-Genome Resequencing of Worldwide Wild and Domestic Sheep Elucidates Genetic Diversity, Introgression, and Agronomically Important Loci. Mol. Biol. Evol. 2022, 39, msab353. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Flori, L.; Riebler, A.; Jaffrézic, F.; Laloé, D.; Gut, I.; Moazami-Goudarzi, K.; Foulley, J.L. A Whole Genome Bayesian Scan for Adaptive Genetic Divergence in West African Cattle. BMC Genom. 2009, 10, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tissot van Patot, M.C.; Gassmann, M. Hypoxia: Adapting to High Altitude by Mutating EPAS-1, the Gene Encoding HIF-2α. High Alt. Med. Biol. 2011, 12, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Sikora, K.M.; Magee, D.A.; Berkowicz, E.W.; Berry, D.P.; Howard, D.J.; Mullen, M.P.; Evans, R.D.; Machugh, D.E.; Spillane, C. DNA Sequence Polymorphisms within the Bovine Guanine Nucleotide-Binding Protein Gs Subunit Alpha (Gsα)-Encoding (GNAS) Genomic Imprinting Domain Are Associated with Performance Traits. BMC Genet. 2011, 12, 4. [Google Scholar] [CrossRef] [Green Version]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional Regulation of Vascular Endothelial Cell Responses to Hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Del Rey, M.J.; Izquierdo, E.; Usategui, A.; Gonzalo, E.; Blanco, F.J.; Acquadro, F.; Pablos, J.L. The Transcriptional Response of Normal and Rheumatoid Arthritis Synovial Fibroblasts to Hypoxia. Arthritis Care Res. 2010, 62, 3584–3594. [Google Scholar] [CrossRef]
- Ortiga-Carvalho, T.M.; Sidhaye, A.R.; Wondisford, F.E. Thyroid Hormone Receptors and Resistance to Thyroid Hormone Disorders. Nat. Rev. Endocrinol. 2014, 10, 582–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, B. Spontaneous T Cell Proliferation: A Physiologic Process to Create and Maintain Homeostatic Balance and Diversity of the Immune System. Front. Immunol. 2018, 9, 547. [Google Scholar] [CrossRef]
- Koyasu, S.; Moro, K. Type 2 Innate Immune Responses and the Natural Helper Cell. Immunology 2011, 132, 475–481. [Google Scholar] [CrossRef]
- Naserkheil, M.; Mehrban, H.; Lee, D.; Park, M.N. Genome-Wide Association Study for Carcass Primal Cut Yields Using Single-Step Bayesian Approach in Hanwoo Cattle. Front. Genet. 2021, 12, 752424. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, V.; Miharada, K. Regulation of Unfolded Protein Response in Hematopoietic Stem Cells. Int. J. Hematol. 2018, 107, 627–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, G.A.; Rodriguez, M.; Kathrein, K.L. Regulation of Stress-Induced Hematopoiesis. Curr. Opin. Hematol. 2020, 27, 279–287. [Google Scholar] [CrossRef]
- Al-Hashem, F.H.; Alkhateeb, M.A.; Shatoor, A.S.; Khalil, M.A.; Sakr, H.F. Chronic Exposure of Rats to Native High Altitude Increases in Blood Pressure via Activation of the Renin-Angiotensin-Aldosterone System. Saudi Med. J. 2012, 33, 1169–1176. [Google Scholar] [CrossRef]
- Bernabucci, U.; Lacetera, N.; Baumgard, L.H.; Rhoads, R.P.; Ronchi, B.; Nardone, A. Metabolic and Hormonal Acclimation to Heat Stress in Domesticated Ruminants. Animal 2010, 4, 1167–1183. [Google Scholar] [CrossRef] [Green Version]
- Weir, E.K.; Olschewski, A. Role of Ion Channels in Acute and Chronic Responses of the Pulmonary Vasculature to Hypoxia. Cardiovasc. Res. 2006, 71, 630–641. [Google Scholar] [CrossRef] [Green Version]
- Makina, S.O.; Muchadeyi, F.C.; Van Marle-Köster, E.; Taylor, J.F.; Makgahlela, M.L.; Maiwashe, A. Genome-Wide Scan for Selection Signatures in Six Cattle Breeds in South Africa. Genet. Sel. Evol. 2015, 47, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuefner, M.S.; Deng, X.; Stephenson, E.J.; Pham, K.; Park, E.A. Secretory Phospholipase A2 Group IIA Enhances the Metabolic Rate and Increases Glucose Utilization in Response to Thyroid Hormone. FASEB J. 2019, 33, 738–749. [Google Scholar] [CrossRef]
- Fontanesi, L.; D’Alessandro, E.; Scotti, E.; Liotta, L.; Crovetti, A.; Chiofalo, V.; Russo, V. Genetic Heterogeneity and Selection Signature at the KIT Gene in Pigs Showing Different Coat Colours and Patterns. Anim. Genet. 2010, 41, 478–492. [Google Scholar] [CrossRef]
- Terefe, E.; Haile, A.; Mulatu, W.; Dessie, T.; Mwai, O. Phenotypic Characteristics and Trypanosome Prevalence of Mursi Cattle Breed in the Bodi and Mursi Districts of South Omo Zone, Southwest Ethiopia. Trop. Anim. Health Prod. 2015, 47, 485–493. [Google Scholar] [CrossRef]
- Lemecha, H.; Mulatu, W.; Hussein, I.; Rege, E.; Tekle, T.; Abdicho, S.; Ayalew, W. Response of Four Indigenous Cattle Breeds to Natural Tsetse and Trypanosomosis Challenge in the Ghibe Valley of Ethiopia. Vet. Parasitol. 2006, 141, 165–176. [Google Scholar] [CrossRef]
- Mauki, D.H.; Adeola, A.C.; Ng’ang’a, S.I.; Tijjani, A.; Akanbi, I.M.; Sanke, O.J.; Abdussamad, A.M.; Olaogun, S.C.; Ibrahim, J.; Dawuda, P.M.; et al. Genetic Variation of Nigerian Cattle Inferred from Maternal and Paternal Genetic Markers. PeerJ 2021, 9, e10607. [Google Scholar] [CrossRef] [PubMed]
- Meseret, S.; Mekonnen, Y.A.; Brenig, B.; Schütz, E.; Hanotte, O.; Gültas, M.; Schmitt, A.O. Genetic Diversity and Population Structure of Six Ethiopian Cattle Breeds from Different Geographical Regions Using High Density Single Nucleotide Polymorphisms. Livest. Sci. 2020, 234, 103979. [Google Scholar] [CrossRef]
- Kim, J.; Hanotte, O.; Mwai, O.A.; Dessie, T.; Bashir, S.; Diallo, B.; Agaba, M.; Kim, K.; Kwak, W.; Sung, S.; et al. The Genome Landscape of Indigenous African Cattle. Genome Biol. 2017, 18, 34. [Google Scholar] [CrossRef]
- Edea, Z.; Dadi, H.; Dessie, T.; Uzzaman, M.R.; Rothschild, M.F.; Kim, E.S.; Sonstegard, T.S.; Kim, K.S. Genome-Wide Scan Reveals Divergent Selection among Taurine and Zebu Cattle Populations from Different Regions. Anim. Genet. 2018, 49, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of Homozygosity and Distribution of Functional Variants in the Cattle Genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and Runs of Homozygosity: A Possible Solution to an Old Problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Forutan, M.; Ansari Mahyari, S.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and Runs of Homozygosity before and after Genomic Selection in North American Holstein Cattle. BMC Genom. 2018, 19, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceballos, F.C.; Gürün, K.; Altınışık, N.E.; Gemici, H.C.; Karamurat, C.; Koptekin, D.; Vural, K.B.; Mapelli, I.; Sağlıcan, E.; Sürer, E.; et al. Human Inbreeding Has Decreased in Time through the Holocene. Curr. Biol. 2021, 31, 3925–3934. [Google Scholar] [CrossRef] [PubMed]
- Qanbari, S.; Pausch, H.; Jansen, S.; Somel, M.; Strom, T.M.; Fries, R.; Nielsen, R.; Simianer, H. Classic Selective Sweeps Revealed by Massive Sequencing in Cattle. PLoS Genet. 2014, 10, e1004148. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, M.; Andreani, V.; Kapoor, T.; Herp, S.; Flach, H.; Duchniewicz, M.; Grosschedl, R. MZB1 Is a GRP94 Cochaperone That Enables Proper Immunoglobulin Heavy Chain Biosynthesis upon ER Stress. Genes Dev. 2014, 28, 1165–1178. [Google Scholar] [CrossRef] [Green Version]
- Guan, C.; Niu, Y.; Chen, S.C.; Kang, Y.; Wu, J.X.; Nishi, K.; Chang, C.C.Y.; Chang, T.Y.; Luo, T.; Chen, L. Structural Insights into the Inhibition Mechanism of Human Sterol O-Acyltransferase 1 by a Competitive Inhibitor. Nat. Commun. 2020, 11, 2478. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [Green Version]
- Kemp, M.G.; Lindsey-Boltz, L.A.; Sancar, A. UV Light Potentiates STING (Stimulator of Interferon Genes)-Dependent Innate Immune Signaling through Deregulation of ULK1 (Unc51-like Kinase 1). J. Biol. Chem. 2015, 290, 12184–12194. [Google Scholar] [CrossRef] [Green Version]
- Wettschureck, N.; Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [Green Version]
- Imumorin, I.G.; Kim, E.H.; Lee, Y.M.; De Koning, D.J.; van Arendonk, J.A.; De Donato, M.; Taylor, J.F.; Kim, J.J. Genome Scan for Parent-of-Origin QTL Effects on Bovine Growth and Carcass Traits. Front. Genet. 2011, 2, 44. [Google Scholar] [CrossRef] [Green Version]
- Taneera, J.; Dhaiban, S.; Mohammed, A.K.; Mukhopadhyay, D.; Aljaibeji, H.; Sulaiman, N.; Fadista, J.; Salehi, A. GNAS Gene Is an Important Regulator of Insulin Secretory Capacity in Pancreatic β-Cells. Gene 2019, 715, 144028. [Google Scholar] [CrossRef]
- Iung, L.H.D.S.; Mulder, H.A.; Neves, H.H.D.R.; Carvalheiro, R. Genomic Regions Underlying Uniformity of Yearling Weight in Nellore Cattle Evaluated under Different Response Variables. BMC Genom. 2018, 19, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes de Oliveira, E.; Keogh, J.M.; Talbot, F.; Henning, E.; Ahmed, R.; Perdikari, A.; Bounds, R.; Wasiluk, N.; Ayinampudi, V.; Barroso, I.; et al. Obesity-Associated GNAS Mutations and the Melanocortin Pathway. New Engl. J. Med. 2021, 385, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tang, J.; Haines, C.J.; Feng, H.; Lai, L.; Teng, X.; Han, Y. C-Kit and Its Related Genes in Spermatogonial Differentiation. Spermatogenesis 2011, 1, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Rossi, P.; Sette, C.; Dolci, S.; Geremia, R. Role of C-Kit in Mammalian Spermatogenesis. J. Endocrinol. Investig. 2000, 23, 609–615. [Google Scholar] [CrossRef] [Green Version]
- Fontanesi, L.; Tazzoli, M.; Russo, V.; Beever, J. Genetic Heterogeneity at the Bovine KIT Gene in Cattle Breeds Carrying Different Putative Alleles at the Spotting Locus. Anim. Genet. 2010, 41, 295–303. [Google Scholar] [CrossRef]
- Hulsman Hanna, L.L.; Sanders, J.O.; Riley, D.G.; Abbey, C.A.; Gill, C.A. Identification of a Major Locus Interacting with MC1R and Modifying Black Coat Color in an F2 Nellore-Angus Population. Genet. Sel. Evol. 2014, 46, 4. [Google Scholar] [CrossRef] [Green Version]
- Haase, B.; Brooks, S.A.; Schlumbaum, A.; Azor, P.J.; Bailey, E.; Alaeddine, F.; Mevissen, M.; Burger, D.; Poncet, P.-A.; Rieder, S.; et al. Allelic Heterogeneity at the Equine KIT Locus in Dominant White (W) Horses. PLoS Genet. 2007, 3, e195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boitard, S.; Boussaha, M.; Capitan, A.; Rocha, D.; Servin, B. Uncovering Adaptation from Sequence Data: Lessons from Genome Resequencing of Four Cattle Breeds. Genetics 2016, 203, 433–450. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.S.W.; Wang, X.; Roberts, S.S.; Griffey, S.M.; Reczek, P.R.; Wolgemuth, D.J. Oral Administration of a Retinoic Acid Receptor Antagonist Reversibly Inhibits Spermatogenesis in Mice. Endocrinology 2011, 152, 2492–2502. [Google Scholar] [CrossRef] [Green Version]
- Peer, N.R.; Law, S.M.; Murdoch, B.; Goulding, E.H.; Eddy, E.M.; Kim, K. Germ Cell–Specific Retinoic Acid Receptor a Functions in Germ Cell Organization, Meiotic Integrity, and Spermatogonia. Endocrinology 2018, 159, 3403–3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, J.; Zhang, L.; Shi, S.; Zhou, X.; Hu, Y.; Gao, L.; Yang, G.; Pang, W.; Chen, H.; et al. NR1D1 Targeting CYP19A1 Inhibits Estrogen Synthesis in Ovarian Granulosa Cells. Theriogenology 2022, 180, 17–29. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, L.; Li, Y.; Dong, H.; Zhang, H.; Zhang, Y.; Ma, T.; Yang, L.; Gao, D.; Wang, X.; et al. Circadian Clock Regulates Granulosa Cell Autophagy through NR1D1-Mediated Inhibition of ATG5. Am. J. Physiol.-Cell Physiol. 2022, 322, C231–C245. [Google Scholar] [CrossRef]
- Dai, L.; Zhang, Q.; Shi, J.; Bai, X.; An, X.; Zhang, B.; Zhang, Y.; Zhao, X. The Distribution, Expression Patterns and Functional Analysis of Nr1d1 and Nr4a2 in the Reproductive Axis Tissues of the Male Tianzhu White Yak. Animals 2021, 11, 3117. [Google Scholar] [CrossRef]
- Li, C.; Zhang, L.; Ma, T.; Gao, L.; Yang, L.; Wu, M.; Pang, Z.; Wang, X.; Yao, Q.; Xiao, Y.; et al. Bisphenol A Attenuates Testosterone Production in Leydig Cells via the Inhibition of NR1D1 Signaling. Chemosphere 2021, 263, 128020. [Google Scholar] [CrossRef]
- Hunter, A.L.; Pelekanou, C.E.; Barron, N.J.; Northeast, R.C.; Grudzien, M.; Adamson, A.D.; Downton, P.; Cornfield, T.; Cunningham, P.S.; Billaud, J.-N.; et al. Adipocyte NR1D1 Dictates Adipose Tissue Expansion during Obesity. Elife 2021, 10, e63324. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liao, F.; Luo, G.; Qian, Y.; He, X.; Xu, W.; Ding, S.; Pu, J. NR1D1 Deletion Induces Rupture-Prone Vulnerable Plaques by Regulating Macrophage Pyroptosis via the NF- κ B/NLRP3 Inflammasome Pathway. Oxidative Med. Cell. Longev. 2021, 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Pariollaud, M.; Gibbs, J.E.; Hopwood, T.W.; Brown, S.; Begley, N.; Vonslow, R.; Poolman, T.; Guo, B.; Saer, B.; Jones, D.H.; et al. Circadian Clock Component REV-ERBα Controls Homeostatic Regulation of Pulmonary Inflammation. J. Clin. Investig. 2018, 128, 2281–2296. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.H.; Holt, T.N.; Hedges, L.K.; Womack, B.; Memon, S.S.; Willers, E.D.; Wheeler, L.; III, J.A.P.; Hamid, R. High-Altitude Pulmonary Hypertension in Cattle (Brisket Disease): Candidate Genes and Gene Expression Profiling of Peripheral Blood Mononuclear Cells. Pulm. Circ. 2011, 1, 462–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remillard, C.V.; Yuan, J.X.-J. High Altitude Pulmonary Hypertension: Role of K+ and Ca2+ Channels. High Alt. Med. Biol. 2005, 6, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular Smooth Muscle Contraction in Hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.G.; Ye, Y. Bag6/Bat3/Scythe: A Novel Chaperone Activity with Diverse Regulatory Functions in Protein Biogenesis and Degradation. BioEssays 2013, 35, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Bracher, A.; Verghese, J. The Nucleotide Exchange Factors of Hsp70 Molecular Chaperones. Front. Mol. Biosci. 2015, 2, 10. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Cattle Population | Region (District) | Altitude (m.a.s.l) | GPS Coordinates | Climate/Agro-Ecology | |
|---|---|---|---|---|---|---|
| Latitude | Longitude | |||||
| 1 | Afar | Afar (Werer/Asayta) | 804 | 9.35 | 40.17 | Hot, arid, low altitude |
| 2 | Arsi | Arsi (Arsi Bekoji) | 2800 | 7.58 | 39.28 | Cold, humid, mid altitude |
| 3 | Bagaria | Benishangul (AL mahal) | 680 | 11.55 | 35.16 | Hot, humid, low altitude |
| 4 | Bale | Bale (Bale Mountain) | 3586 | 6.77 | 39.76 | Cold, humid, high altitude |
| 5 | Begait | Gonder (Humera) | 895 | 14.10 | 37.22 | Hot, humid, low altitude |
| 6 | Boran | Borena (Borena plain) | 1368 | 4.55 | 38.10 | Hot, humid, low altitude |
| 7 | Choke | Gojam (Choke Mountain) | 3410 | 10.61 | 37.84 | Cold, humid, high altitude |
| 8 | Fogera | Bahir Dar (Fogera plain) | 1735 | 11.30 | 37.28 | Cold, humid, mid altitude |
| 9 | Goffa | Goffa (Arba Minch) | 1100 | 6.21 | 37.07 | Hot, humid, low altitude |
| 10 | Horro | Welega (Horro) | 1722 | 9.07 | 37.03 | Cold, humid, mid altitude |
| 11 | Mursi | South Omo (Mursi) | 1405 | 5.47 | 36.34 | Hot, humid, low altitude |
| 12 | Ogaden | Somali (Kebri-Beyah) | 1200 | 9.59 | 41.86 | Hot, arid, low altitude |
| 13 | Semien | Gonder (Semien Mountain) | 3732 | 13.24 | 38.14 | Cold, humid, high altitude |
| 14 | Sheko | Kaffa Shaka | 2240 | 7.65 | 35.51 | Hot, humid, mid altitude |
| ROHs ≥ 0.5 Mb | ROHs ≥ 1.0 Mb | Nucleotide Diversity | ||||||
|---|---|---|---|---|---|---|---|---|
| Breed | N | No. | Froh | No. | Froh | E(Het) | O(Het) | |
| Afar | 14 | 416 | 0.0132 | 43 | 0.0024 | 3.49 × 10−3 | 0.279 | 0.273 |
| Arsi | 10 | 731 | 0.0224 | 107 | 0.0059 | 3.44 × 10−3 | 0.279 | 0.259 |
| Bagaria | 10 | 553 | 0.0157 | 57 | 0.0029 | 3.59 × 10−3 | 0.280 | 0.286 |
| Bale | 10 | 270 | 0.008 | 17 | 0.0014 | 3.61× 10−3 | 0.280 | 0.291 |
| Begait | 9 | 468 | 0.0197 | 55 | 0.0048 | 3.45 × 10−3 | 0.279 | 0.264 |
| Boran | 10 | 460 | 0.0124 | 32 | 0.0017 | 3.52 × 10−3 | 0.279 | 0.260 |
| Choke | 10 | 216 | 0.0069 | 7 | 0.0005 | 3.60 × 10−3 | 0.280 | 0.289 |
| Fogera | 12 | 695 | 0.0247 | 128 | 0.008 | 3.15 × 10−3 | 0.278 | 0.241 |
| Goffa | 13 | 462 | 0.0137 | 66 | 0.0035 | 3.11 × 10−3 | 0.279 | 0.242 |
| Horro | 11 | 681 | 0.0184 | 84 | 0.0051 | 3.38 × 10−3 | 0.279 | 0.254 |
| Mursi | 10 | 616 | 0.0202 | 70 | 0.0043 | 3.32 × 10−3 | 0.279 | 0.252 |
| Ogaden | 9 | 663 | 0.0316 | 142 | 0.0119 | 2.88 × 10−3 | 0.275 | 0.193 |
| Semien | 10 | 167 | 0.0054 | 6 | 0.0008 | 3.64 × 10−3 | 0.280 | 0.294 |
| Sheko | 13 | 622 | 0.0204 | 87 | 0.005 | 2.67 × 10−3 | 0.278 | 0.217 |
| Butana | 10 | 628 | 0.0225 | 129 | 0.0079 | 3.30 × 10−3 | 0.279 | 0.264 |
| Kenana | 10 | 1008 | 0.0347 | 248 | 0.0145 | 3.06 × 10−3 | 0.277 | 0.214 |
| Ankole | 10 | 763 | 0.0229 | 86 | 0.0057 | 2.39 × 10−3 | 0.276 | 0.177 |
| Muturu | 10 | 3864 | 0.119 | 596 | 0.0319 | 1.26 × 10−3 | 0.278 | 0.075 |
| N’Dama | 10 | 2200 | 0.0931 | 654 | 0.0525 | 2.26 × 10−3 | 0.277 | 0.125 |
| Gir | 9 | 1087 | 0.0355 | 220 | 0.0126 | 2.41 × 10−3 | 0.275 | 0.178 |
| Angus | 10 | 2762 | 0.129 | 1112 | 0.085 | 1.39 × 10−3 | 0.279 | 0.121 |
| Holstein | 11 | 2325 | 0.1024 | 847 | 0.0627 | 1.43 × 10−3 | 0.279 | 0.130 |
| Breed | ANG | AFR | ANK | ARS | BAG | BAL | BEG | BOR | BUT | CHO | FOG | GIR | GOF | HOL | HOR | KEN | MUR | MUT | NDA | OGD | SEM |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ANG | |||||||||||||||||||||
| AFR | 0.308 | ||||||||||||||||||||
| ANK | 0.248 | 0.092 | |||||||||||||||||||
| ARS | 0.306 | 0.010 | 0.075 | ||||||||||||||||||
| BAG | 0.323 | 0.019 | 0.063 | 0.019 | |||||||||||||||||
| BAL | 0.312 | 0.012 | 0.086 | 0.006 | 0.017 | ||||||||||||||||
| BEG | 0.282 | 0.019 | 0.091 | 0.018 | 0.013 | 0.021 | |||||||||||||||
| BOR | 0.289 | 0.010 | 0.087 | 0.008 | 0.019 | 0.008 | 0.021 | ||||||||||||||
| BUT | 0.329 | 0.049 | 0.119 | 0.046 | 0.045 | 0.053 | 0.038 | 0.049 | |||||||||||||
| CHO | 0.303 | 0.009 | 0.079 | 0.003 | 0.014 | 0.003 | 0.016 | 0.008 | 0.048 | ||||||||||||
| FOG | 0.293 | 0.009 | 0.072 | 0.002 | 0.017 | 0.009 | 0.018 | 0.013 | 0.045 | 0.004 | |||||||||||
| GIR | 0.439 | 0.009 | 0.205 | 0.086 | 0.087 | 0.089 | 0.084 | 0.083 | 0.112 | 0.087 | 0.087 | ||||||||||
| GOF | 0.289 | 0.009 | 0.070 | 0.006 | 0.024 | 0.013 | 0.025 | 0.013 | 0.049 | 0.009 | 0.009 | 0.095 | |||||||||
| HOL | 0.122 | 0.009 | 0.250 | 0.308 | 0.325 | 0.314 | 0.284 | 0.291 | 0.331 | 0.304 | 0.296 | 0.442 | 0.292 | ||||||||
| HOR | 0.292 | 0.009 | 0.068 | 0.001 | 0.020 | 0.009 | 0.020 | 0.012 | 0.047 | 0.004 | 0.002 | 0.092 | 0.007 | 0.294 | |||||||
| KEN | 0.297 | 0.009 | 0.084 | 0.022 | 0.022 | 0.031 | 0.015 | 0.031 | 0.046 | 0.026 | 0.021 | 0.095 | 0.023 | 0.299 | 0.022 | ||||||
| MUR | 0.277 | 0.009 | 0.056 | 0.009 | 0.029 | 0.018 | 0.029 | 0.018 | 0.056 | 0.014 | 0.011 | 0.112 | 0.008 | 0.279 | 0.008 | 0.029 | |||||
| MUT | 0.320 | 0.009 | 0.268 | 0.332 | 0.346 | 0.335 | 0.299 | 0.308 | 0.364 | 0.327 | 0.317 | 0.479 | 0.311 | 0.309 | 0.315 | 0.336 | 0.297 | ||||
| NDA | 0.264 | 0.009 | 0.189 | 0.245 | 0.258 | 0.252 | 0.229 | 0.238 | 0.234 | 0.244 | 0.235 | 0.387 | 0.231 | 0.262 | 0.231 | 0.092 | 0.212 | 0.234 | |||
| OGA | 0.312 | 0.009 | 0.073 | 0.003 | 0.029 | 0.020 | 0.026 | 0.015 | 0.054 | 0.016 | 0.012 | 0.095 | 0.013 | 0.317 | 0.012 | 0.023 | 0.007 | 0.348 | 0.252 | ||
| SEM | 0.307 | 0.009 | 0.084 | 0.006 | 0.013 | 0.005 | 0.016 | 0.010 | 0.047 | 0.001 | 0.005 | 0.086 | 0.012 | 0.308 | 0.007 | 0.027 | 0.018 | 0.329 | 0.247 | 0.020 | |
| SHE | 0.251 | 0.009 | 0.048 | 0.027 | 0.047 | 0.034 | 0.044 | 0.039 | 0.074 | 0.029 | 0.029 | 0.139 | 0.023 | 0.252 | 0.023 | 0.044 | 0.021 | 0.267 | 0.060 | 0.034 | 0.032 |
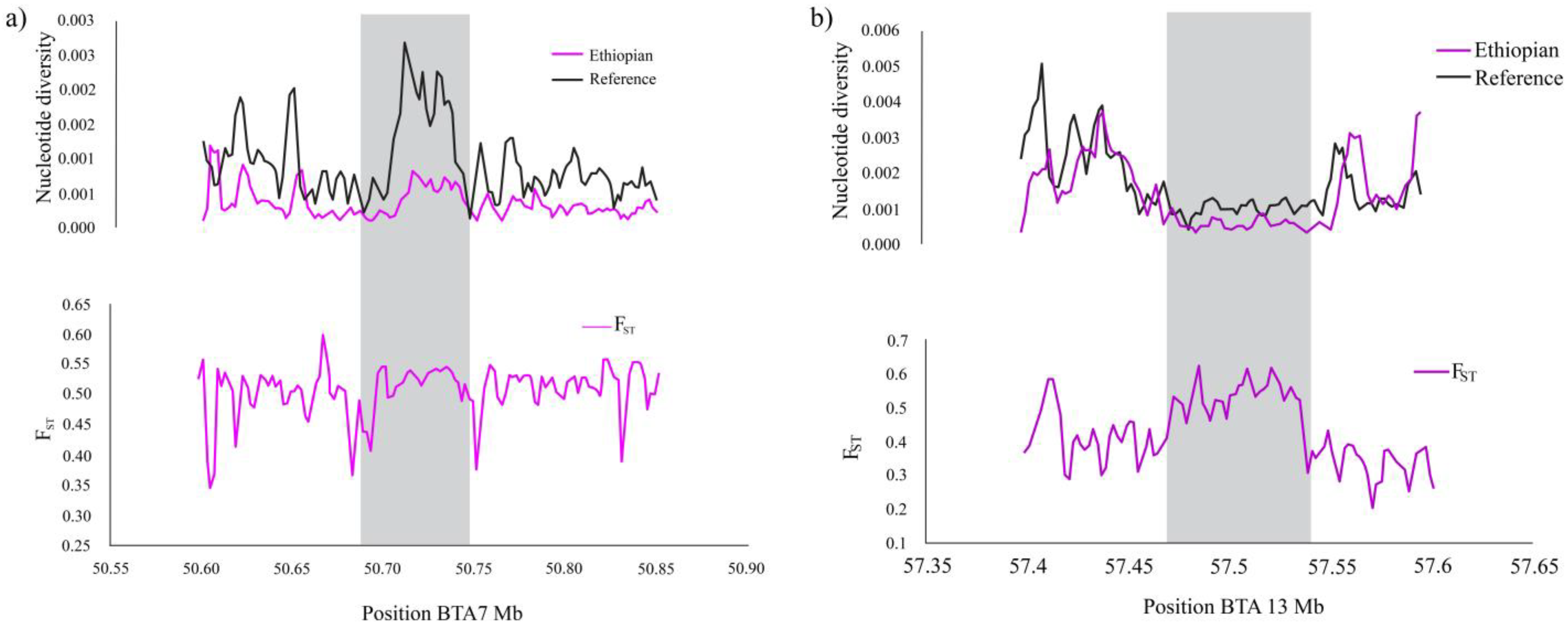
| BTA | Region Start | Region Stop | Genes Name | FST | XP-CLR | ZHp | iHS | References |
|---|---|---|---|---|---|---|---|---|
| 5 | 48.65 | 48.75 | WIF1 | 5.78 | 93 | −3.65 | - | [48] |
| 58.55 | 58.65 | OR6C75, OR6C1Q | 5.10 | 31 | - | 5.86 | [49,50] | |
| 112.35 | 112.45 | EP300, L3MBTL2, ZC3H7B, CHADL, RANGAP1 | 4.29 | 35 | - | 5.70 | [50,51] | |
| 7 | 50.00 | 50.10 | ENSBTAG00000004415 | 4.53 | 102 | −3.96 | - | [17,52,53] |
| 50.15 | 50.25 | CTNNA1, LRRTM2 | 5.21 | 167 | −4.54 | - | [17,54] | |
| 50.25 | 50.35 | SIL1 | 5.77 | 186 | −3.56 | - | [17,55] | |
| 50.60 | 50.70 | MATR3, PAIP2 | 5.69 | 80 | −4.39 | - | [17,45,56] | |
| 50.65 | 50.75 | SLC23A1, MZB1, PROB1, SPATA24, DNAJC18, ECSCR, SMIM33, STING1 | 6.02 | 287 | −4.39 | - | [17,53,57] | |
| 50.85 | 50.95 | UBE2D2, CXXC5 | 6.22 | 177 | −4.70 | - | [17,45] | |
| 51.0 | 51.1 | PSD2, NRG2 | 5.52 | 336 | −4.34 | - | [17] | |
| 13 | 57.4 | 57.5 | ENSBTAG00000017475, GNAS | 4.11 | 37 | −4.05 | 5.09 | [17,58] |
| NELFCD | 4.11 | 37 | - | 5.09 | ||||
| 16 | 19.5 | 19.6 | USH2A | - | 56 | −4.09 | 6.28 | |
| 19 | 39.65 | 39.75 | FBXL20 | 4.48 | 40 | −3.89 | - | [17] |
| 39.75 | 39.85 | CDK12, MED1 | 3.97 | 125 | −3.89 | - | [17,59] | |
| 40.4 | 40.5 | CASC3, RAPGEFL1, WIPF2 | 5.50 | 148 | −3.63 | 6.37 | [56,60] | |
| MSL1 | 5.50 | - | −3.63 | 6.37 | ||||
| 40.3 | 40.4 | THRA, MED24, NR1D1 | 4.54 | 43 | - | 6.37 | [61] |
| Term | Count | p Value | Fold Enrichment | Genes |
|---|---|---|---|---|
| GO:0001501~skeletal system development | 46 | 1.2× 10−7 | 2.4 | FGF18, KIT, RAB33B, HOXC10, HOXC4, HOXD13, HOXD12, HOXD3, IRX5, HOXB7, HOXC9, BMPR2, PDGFRA, HOXC6, ZFAND5, HOXB2, GSC, THRA, FGR, CYP26B1, RARA, ADAMTS12, MED1, MPIG6B, EDNRA, BBS2, HOXD10, HOXD9, EP300, EFEMP1, TIFAB, HOXB3, ACTN3, LHX1, HOXD4, HOXB4, CHADL, HMGA2, HOXB5, HOXD8, HOXB6, GNAS, LY6G6D, HOXB8, WNT5B, HOXB9 |
| GO:0002699~positive regulation of immune effector process | 16 | 2.1 × 10−2 | 1.9 | IL18, IL18R1, KIT, IL23A, SPHK2, STAT5B, FGR, RARA, PRKCZ, TBX21, IL13, IL4, EXOSC3, IL18RAP, MZB1, TRIM6 |
| GO:0002292~T cell differentiation involved in immune response | 9 | 5.1 × 10−3 | 3.3 | IL18, IL18R1, IL23A, RARA, PRKCZ, TBX21, RORA, IL4, LOXL3 |
| GO:0050778~positive regulation of immune response | 38 | 6.8 × 10−3 | 1.6 | IL18, MYD88, IL18R1, STING1, HMCN2, KIT, IL23A, POLR3B, IRAK3, SPHK2, STAT5B, NR1H3, RTN4, FGR, NR1D1, RARA, MED1, PRKCZ, TBX21, CCR7, IL13, IL4, EXOSC3, BAG6, OTUD4, IL18RAP, ALPK1, CHADL, PAWR, CRKL, ELANE, TNIP3, CFD, TRIM6 |
| GO:0042092~type 2 immune response | 6 | 8.4 × 10−3 | 4.6 | IL18, TRAF3IP2, PRKCZ, TBX21, IL4 |
| GO:0019915~lipid storage | 9 | 9.3 × 10−3 | 3.0 | SOAT1, STAT5A, STAT5B, NR1H3, CD36, FAM71F2, PLIN5, EHD1 |
| GO:0071218~cellular response to misfolded protein | 5 | 1.9 × 10−2 | 4.7 | DNAJC18, RNF126, BAG6, AUP1, RNF5 |
| GO:1903706~regulation of hemopoiesis | 22 | 5.0 × 10−2 | 1.5 | IL18, FSTL3, MAFB, IL23A, STAT5A, STAT5B, NUDT21, HSPA9, CYP26B1, RARA, LOX, MED1, PRKCZ, TBX21, AGER, TOB2, CASP8, YTHDF2, IL4, LOXL3, CSF3, HOXB8 |
| GO:0005254~chloride channel activity | 9 | 1.8 × 10−2 | 2.7 | PACC1, ANO6, CLCA1, GABRB3, CLIC1, CLCA4, GLRB, ANO3, CLCA2 |
| bta04915:Estrogen signaling pathway | 18 | 3.5 × 10−4 | 2.7 | TGFA, ITPR2, KRT20, ATF6B, PRKACB, RARA, KRT12, SHC2, KRT40, KRT39, KRT10, SHC4, KRT25, KRT26, KRT27, KRT28, GRM1, GNAS |
| bta04975:Fat digestion and absorption | 9 | 3.3× 10−3 | 3.6 | PLPP2, AGPAT1, PLA2G2A, CD36, PLA2G5 |
| bta04972:Pancreatic secretion | 13 | 3.7 × 10−3 | 2.6 | ITPR2, PLA2G2A, CLCA1, ATP2B1, CCK, CLCA4, CLCA2, PLA2G5, GNAS |
| bta04270:Vascular smooth muscle contraction | 13 | 3.9 × 10−2 | 1.9 | ITPR2, PLA2G2A, PRKACB, EDNRA, PLA2G5, GUCY1A2, GNAS |
| BTA | SNP Id | Position and Type of Allele Substitution | Gene | Amino Acid Change and Position | Exon | Codon Change | Frequency |
|---|---|---|---|---|---|---|---|
| 5 | rs482168794 | g.83460939T>C | ITPR2 | P: M2195T | 49/57 | aTg/aCg | 0.372 |
| 5 | rs516675337 | g.112389173C>G | CHADL | P: V445L | 3/5 | Gtg/Ctg | 0.725 |
| 5 | rs520244098 | g.112389376T>C | CHADL | P: D377G | 3/5 | gAc/gGc | 0.812 |
| 5 | rs714095032 | g.112389392G>C | CHADL | P: P372A | 3/5 | Ccc/Gcc | 0.107 |
| 6 | rs714949670 | g.70205294A>G | KIT | P: D60G | 2/21 | gAt/gGt | 0.217 |
| 6 | rs109630427 | g.70214244T>C | KIT | P: M258T | 5/21 | aTg/aCg | 0.939 |
| 7 | rs520476700 | g.50739789G>T | STING1 | P: L201I | 6/8 | Ctc/Atc | 0.928 |
| 13 | rs211162390 | g.57531848A>G | GNAS | P: V144A | 1/13 | gTg/gCg | 0.894 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terefe, E.; Belay, G.; Tijjani, A.; Han, J.; Hanotte, O. Whole Genome Resequencing Reveals Genetic Diversity and Selection Signatures of Ethiopian Indigenous Cattle Adapted to Local Environments. Diversity 2023, 15, 540. https://doi.org/10.3390/d15040540
Terefe E, Belay G, Tijjani A, Han J, Hanotte O. Whole Genome Resequencing Reveals Genetic Diversity and Selection Signatures of Ethiopian Indigenous Cattle Adapted to Local Environments. Diversity. 2023; 15(4):540. https://doi.org/10.3390/d15040540
Chicago/Turabian StyleTerefe, Endashaw, Gurja Belay, Abdulfatai Tijjani, Jianlin Han, and Olivier Hanotte. 2023. "Whole Genome Resequencing Reveals Genetic Diversity and Selection Signatures of Ethiopian Indigenous Cattle Adapted to Local Environments" Diversity 15, no. 4: 540. https://doi.org/10.3390/d15040540
APA StyleTerefe, E., Belay, G., Tijjani, A., Han, J., & Hanotte, O. (2023). Whole Genome Resequencing Reveals Genetic Diversity and Selection Signatures of Ethiopian Indigenous Cattle Adapted to Local Environments. Diversity, 15(4), 540. https://doi.org/10.3390/d15040540