16S rRNA Gene-Based Metagenomic Analysis of Ozark Cave Bacteria

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cave Description and Sample Collection
2.2. DNA Extraction and DNA Sequence Analysis
2.3. Bioinformatics and Statistical Analysis
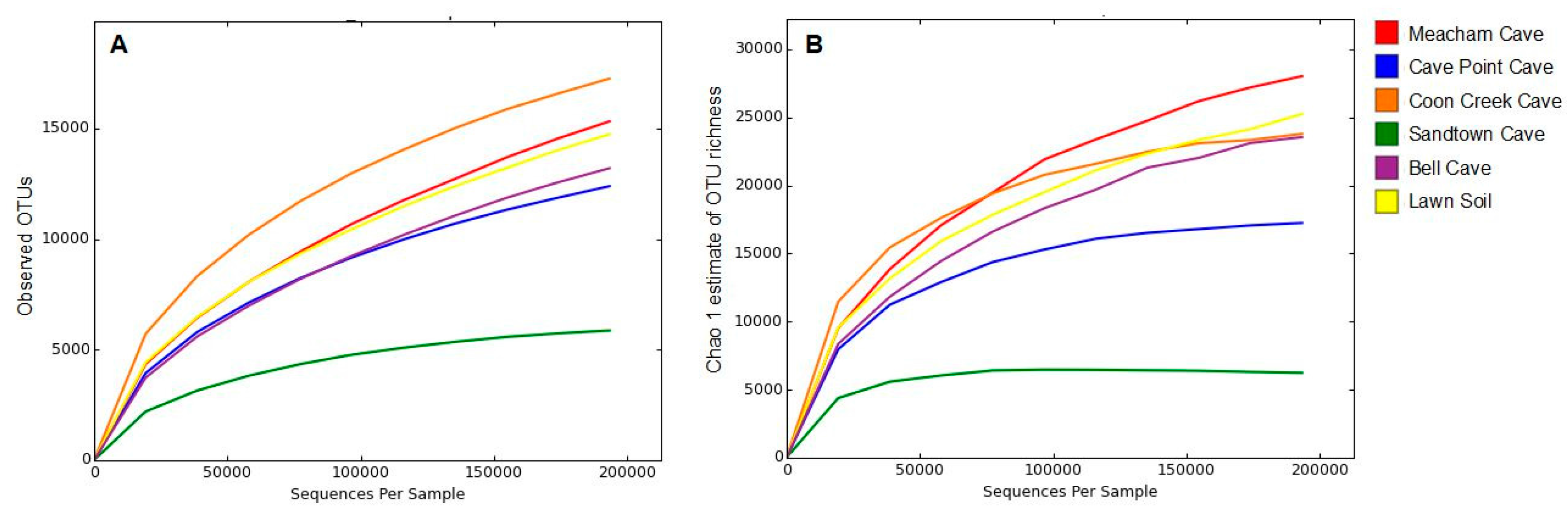
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moore, G.W.; Sullivan, G.N. Speleology: Caves and the Cave Environment, Rev. 3rd ed.; Cave Books: St. Louis, MO, USA, 1997; p. 176. [Google Scholar]
- Barton, H.A.; Jurado, V. What’s up down there? Microbial diversity in caves. Microbe 2007, 2, 132–138. [Google Scholar]
- Barton, H.A. Introduction to cave microbiology: A review for the non-specialist. J. Cave Karst Stud. 2006, 68, 43–54. [Google Scholar]
- Romero, A. Cave Biology: Life in Darkness; Cambridge University Press: New York, NY, USA, 2009; p. 291. [Google Scholar]
- Marshall Hathaway, J.J.; Garcia, M.G.; Balasch, M.M.; Spilde, M.N.; Stone, F.D.; Dapkevicius, M.D.L.N.E.; Amorim, I.R.; Gabriel, R.; Borges, P.A.V.; Northup, D.E. Comparison of bacterial diversity in Azorean and Hawai’ian lava cave microbial mats. Geomicrobiol. J. 2014, 31, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.; Legatzki, A.; Neilson, J.W.; Fryslie, B.; Nelson, W.M.; Wing, R.A.; Soderlund, C.A.; Pryor, B.M.; Maier, R.M. Making a living while starving in the dark: Metagenomic insights into the energy dynamics of a carbonate cave. ISME J. 2014, 8, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T. Lithoautotrophy in the subsurface. FEMS Microbiol. Rev. 1997, 20, 327–337. [Google Scholar] [CrossRef]
- Vlasceanu, L.; Popa, R.; Kinkle, B.K. Characterization of Thiobacillus. thioparus lv43 and its distribution in a chemoautotrophically based groundwater ecosystem. Appl. Environ. Microbiol. 1997, 63, 3123–3127. [Google Scholar] [PubMed]
- Fliermans, C.B.; Bohlool, B.B.; Schmidt, E.L. Autecological study of the chemoautotroph Nitrobacter. by immunofluorescence. Appl. Microbiol. 1974, 27, 124–129. [Google Scholar] [PubMed]
- Northup, D.E.; Barns, S.M.; Yu, L.E.; Spilde, M.N.; Schelble, R.T.; Dano, K.E.; Crossey, L.J.; Connolly, C.A.; Boston, P.J.; Natvig, D.O.; et al. Diverse microbial communities inhabiting ferromanganese deposits in lechuguilla and spider caves. Environ. Microbiol. 2003, 5, 1071–1086. [Google Scholar] [CrossRef] [PubMed]
- Pacton, M.; Breitenbach, S.F.M.; Lechleitner, F.A.; Vaks, A.; Rollion-Bard, C.; Gutareva, O.S.; Osintcev, A.V.; Vasconcelos, C. The role of microorganisms in the formation of a stalactite in Botovskaya Cave, Siberia–paleoenvironmental implications. Biogeosciences 2013, 10, 6115–6130. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauberb, C.L.; Owense, S.; Gilberte, J.A.; Wallh, D.H.; Caporasoe, J.G. Cross–biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [PubMed]
- Staley, J.T.; Konopka, A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu. Rev. Microbiol. 1985, 39, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.J. Growing unculturable bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Pace, N.; Stahl, D.; Lane, D.; Olsen, G. The analysis of natural microbial populations by rRNA sequences. Adv. Microb. Ecol. 1986, 9, 1–55. [Google Scholar]
- Achtman, M.; Wagner, M. Microbial diversity and the genetic nature of microbial species. Nat. Rev. Microbiol. 2008, 6, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glockner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16s rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Mora, C.; Tittensor, D.P.; Adl, S.; Simpson, A.G.B.; Worm, B. How many species are there on earth and in the ocean? PLoS Biol. 2011, 9, e1001127. [Google Scholar] [CrossRef] [PubMed]
- Curtis, T.P.; Sloan, W.T. Exploring microbial diversity—A vast below. Science 2005, 309, 1331–1333. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.H.; Elshahed, M.S. Diversity rankings among bacterial lineages in soil. ISME J. 2008, 3, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Graening, G.O.; Fenolio, D.B.; Slay, M.E. Cave Life of Oklahoma and Arkansas: Exploration and Conservation of Subterranean Biodiversity; University of Oklahoma Press: Norman, OK, USA, 2011; p. 226. [Google Scholar]
- United States Geological Survey. The Geological Map of Arkansas, Digital Version. Available online: http://www.geology.ar.gov/ark_state_maps/Geologic%20Map%20of%20Arkansas%201993%20(34x52) (accessed on 26 July 2017).
- Thomas, D.J.; Boyd, M.; Crowell, K.M.; Curtwright, A.E.; Foll, M.N.; Kuehl, M.M.; McQueen, C.M.; Middaugh, R.; Moore, V.M.; Moreno, M.; et al. A biological inventory of Meacham Cave (independence county, Arkansas). J. Ark. Acad. Sci. 2012, 66, 126–132. [Google Scholar]
- Jones, C.; Dale, M. A Guide to Responsible Caving, 4th ed.; National Speleological Society: Huntsville, AL, USA, 2009; p. 24. [Google Scholar]
- Barron, S.K.; Murdock, C.A.; Blair, B.G.; Meade, M.E.; Barger, T.W. Analysis of bacterial diversity in soils from Blowing Spring Cave (Lauderdale County, AL). J. Ala. Acad. Sci. 2010, 81, 1–10. [Google Scholar]
- Upchurch, R.; Chiu, C.-Y.; Everett, K.; Dyszynski, G.; Coleman, D.C.; Whitman, W.B. Differences in the composition and diversity of bacterial communities from agricultural and forest soils. Soil Biol. Biochem. 2008, 40, 1294–1305. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16s ribosomal rna gene pcr primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Qiime allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16s rRNA gene database and workbench compatible with arb. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. Pynast: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Hamady, M.; Lozupone, C.; Knight, R. Fast unifrac: Facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and phylochip data. ISME J. 2010, 4, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. Unifrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Takacs-Vesbach, C.D. Microbial community analysis of PH 4 thermal springs in Yellowstone National park. Extremophiles 2017, 21, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, D.J.; Wolf, C.R.; Colman, D.R.; Jiang, X.; Kohler, T.J.; McKnight, D.M.; Stanish, L.F.; Yazzie, T.; Takacs-Vesbach, C.D. Patterns of bacterial biodiversity in the glacial meltwater streams of the Mcmurdo Dry Valleys, Antarctica. FEMS Microbiol. Ecol. 2016, 92, fiw148. [Google Scholar] [CrossRef] [PubMed]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Bent, S.J.; Forney, L.J. The tragedy of the uncommon: Understanding limitations in the analysis of microbial diversity. ISME J. 2008, 2, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Lücker, S.; Wagner, M.; Maixner, F.; Pelletier, E.; Koch, H.; Vacherie, B.; Rattei, T.; Damsté, J.S.; Spieck, E.; Le Paslier, D. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc. Natl. Acad. Sci. USA 2010, 107, 13479–13484. [Google Scholar] [CrossRef] [PubMed]
- Ghiorse, W.C.; Hirsch, P. An ultrastructural study of iron and manganese deposition associated with extracellular polymers of Pedomicrobium-like budding bacteria. Arch. Microbiol. 1979, 123, 213–226. [Google Scholar] [CrossRef]
- Peck, S.B. Bacterial deposition of iron, and manganese oxides in north american caves. Natl. Speleol. Soc. Bull. 1986, 48, 26–30. [Google Scholar]
- Nealson, K.H.; Saffarini, D. Iron and manganese in anaerobic respiration: Environmental significance, physiology, and regulation. Annu. Rev. Microbiol. 1994, 48, 311–343. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Shergill, J.K.; Lu, W.P.; Wood, A.P. Oxidative metabolism of inorganic sulfur compounds by bacteria. Antonie Van Leeuwenhoek 1997, 71, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Kamagata, Y.; Oshima, K.; Hanada, S.; Tamaki, H.; Marumo, K.; Maeda, H.; Nedachi, M.; Hattori, M.; Iwasaki, W. Methylocaldum marinum sp. Nov., a thermotolerant, methane-oxidizing bacterium isolated from marine sediments, and emended description of the genus methylocaldum. Int. J. Syst. Evol. Microbiol. 2014, 64, 3240–3246. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.G.; Marina, C.F.; Williams, T. The naturally derived insecticide spinosad is highly toxic to Aedes and Anopheles mosquito larvae. Med. Vet. Entomol. 2004, 18, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Demain, A.L. Pharmaceutically active secondary metabolites of microorganisms. Appl. Microbiol. Biotechnol. 1999, 52, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16s rRNA and 16s rRNA genes. Appl. Microbiol. Biotechnol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Ivarsoon, M.; Holm, N.G. Microbial colonization of various habitable niches during alteration of ocean crust. In Links between Geological Processes, Microbial Activities and Evolution of Life; Springer: Dordrecht, The Netherlands, 2008; Volume 4, pp. 69–111. [Google Scholar]
- Ludwig, W.; Bauer, S.H.; Bauer, M.; Held, I.; Kirchhof, G.; Schulze, R.; Huber, I.; Spring, S.; Hartmann, A.; Schleifer, K.H. Detection and in situ identification of representatives of a widely distributed new bacterial phylum. FEMS Microbiol. Lett. 1997, 153, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Kielak, A.M.; Barreto, C.C.; Kowalchuk, G.A.; van Veen, J.A.; Kuramae, E.E. The ecology of acidobacteria: Moving beyond genes and genomes. Front. Microbiol. 2016, 7, 744. [Google Scholar] [CrossRef] [PubMed]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Klenk, H.P.; Clément, C.; Ouhdouch, Y.; van Wezel, G.P. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Cavaletti, L.; Monciardini, P.; Bamonte, R.; Schumann, P.; Rohde, M.; Sosio, M.; Donadio, S. New lineage of filamentous, spore-forming, gram-positive bacteria from soil. Appl. Microbiol. Biotechnol. 2006, 72, 4360–4369. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, K.H.; Northup, D.E. Bacteria as Indicators of Human Impact in Caves. 7th National Cave and Karst Management Symposium, Proceedings; NICKMS Steering Committee: Albany, NY, USA, 2006; pp. 40–47. [Google Scholar]
- Engel, A.S. Microbial diversity of cave ecosystems. In Geomicrobiology: Molecular and Environmental Perspective; Barton, L.L., Mandl, M., Loy, A., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 219–238. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Cave Entrance Elevation (m) | Cave Temp. (°C) | Cave pH | Total Reads | Reads Passing Quality Filtering | % Reads Passing Quality Filtering | Aligned Merged Reads per Sample |
|---|---|---|---|---|---|---|---|
| Bell Cave | 102 | 13.5 ± 0.5 | 6.5 ± 0.5 | 7,325,566 | 6,394,842 | 87.3% | 990,292 |
| Cave Point Cave | 315 | 13.5 ± 0.5 | 6.5 ± 0.5 | 2,432,496 | 2,120,460 | 87.2% | 292,934 |
| Coon Creek Cave | 159 | 13.5 ± 0.5 | 6.5 ± 0.5 | 2,592,318 | 2,225,312 | 85.8% | 350,957 |
| Meacham Cave | 161 | 13.5 ± 0.5 | 6.5 ± 0.5 | 7,492,255 | 6,438,011 | 85.9% | 865,302 |
| Sandtown Cave | 158 | 13.5 ± 0.5 | 6.5 ± 0.5 | 3,082,676 | 2,682,596 | 87.0% | 193,424 |
| Lawn Soil | NA | NA | NA | 8,208,663 | 6,959,192 | 84.8% | 918,247 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, C.; Gunderman, L.; Coles, C.A.; Lochmann, J.; Parks, M.; Ballard, E.; Glazko, G.; Rahmatallah, Y.; Tackett, A.J.; Thomas, D.J. 16S rRNA Gene-Based Metagenomic Analysis of Ozark Cave Bacteria. Diversity 2017, 9, 31. https://doi.org/10.3390/d9030031
Oliveira C, Gunderman L, Coles CA, Lochmann J, Parks M, Ballard E, Glazko G, Rahmatallah Y, Tackett AJ, Thomas DJ. 16S rRNA Gene-Based Metagenomic Analysis of Ozark Cave Bacteria. Diversity. 2017; 9(3):31. https://doi.org/10.3390/d9030031
Chicago/Turabian StyleOliveira, Cássia, Lauren Gunderman, Cathryn A. Coles, Jason Lochmann, Megan Parks, Ethan Ballard, Galina Glazko, Yasir Rahmatallah, Alan J. Tackett, and David J. Thomas. 2017. "16S rRNA Gene-Based Metagenomic Analysis of Ozark Cave Bacteria" Diversity 9, no. 3: 31. https://doi.org/10.3390/d9030031
APA StyleOliveira, C., Gunderman, L., Coles, C. A., Lochmann, J., Parks, M., Ballard, E., Glazko, G., Rahmatallah, Y., Tackett, A. J., & Thomas, D. J. (2017). 16S rRNA Gene-Based Metagenomic Analysis of Ozark Cave Bacteria. Diversity, 9(3), 31. https://doi.org/10.3390/d9030031