
Interferon (IFN) and Cellular Immune Response Evoked in RNA-Pattern Sensing During Infection with Hepatitis C Virus (HCV)


 and
and
Abstract
:1. Introduction
2. Hydrodynamic Injection of HCV RNA in Mice
3. Extrinsically Transfected RNA Stimulates the MAVS Pathway

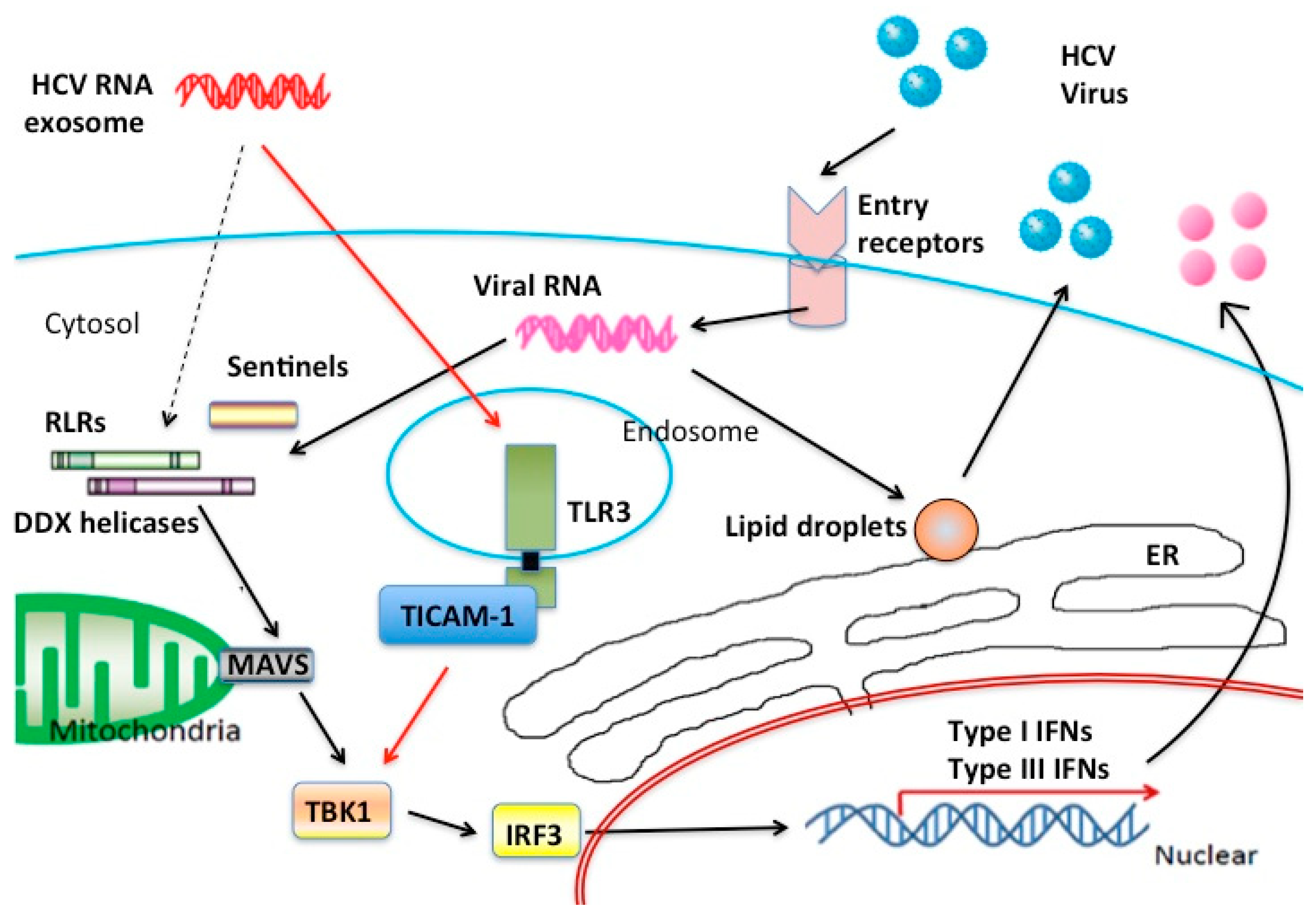{kind=link}
{kind=link}
| Pattern-Recognition Receptor | Adaptor | Natural Agonist | Synthtic Agonist | Pathogen |
|---|---|---|---|---|
| TLR3 | TICAM-1 | Endosomal dsRNA | poly(I:C) | DNA/RNA virus |
| TLR7/8 | MyD88 | Endosomal ssRNA | Imidazoquinoline | RNA virus, bacteria, fungi |
| TLR9 | MyD88 | Non-methylated CpG DNA | CpG ODNs | DNA virus, bacteria |
| RIG-I | MAVS | Cytosolic 5'-ppp-dsRNA | short poly(I:C) | RNA virus, DNA virus |
| MDA5 | MAVS | Cytosolic long dsRNA | long poly(I:C) | RNA virus, bacteria |
| NOD2 | MAVS | Cytosolic ssRNA | muramyl dipeptide | RNA virus, bacteria |
| DDX3 | MAVS | Cytosolic ssRNA, dsRNA | poly(I:C) | RNA virus |
| DDX1/21, DHX36 | TICAM-1 | Cytosolic dsRNA | poly(I:C) | RNA virus |
| DDX41 | STING | Cytosolic dsDNA | - | DNA virus, bacteria |
| DDX60 | MAVS | Cytosolic RNA, dsDNA, | poly(I:C) | DNA/RNA virus |
| DHX9/DHX36 | MyD88 (MAVS) | Cytosolic dsDNA (dsRNA) | CpG ODNs (poly(I:C) | DNA virus |
| DAI (ZBP1) | STING? | Cytosolic dsDNA | - | Dna virus, bacteria |
| NLRP3 | ASC | Cytosolic RNA | sillica, asbestos, alum | DNA/RNA virus, bacteria |
| IFI16 | STING | Cytosolic dsDNA | - | DNA virus |
| LRRFIP1 | β-Catenin | Cytosolic dsDNA | - | DNA virus, bacteria |

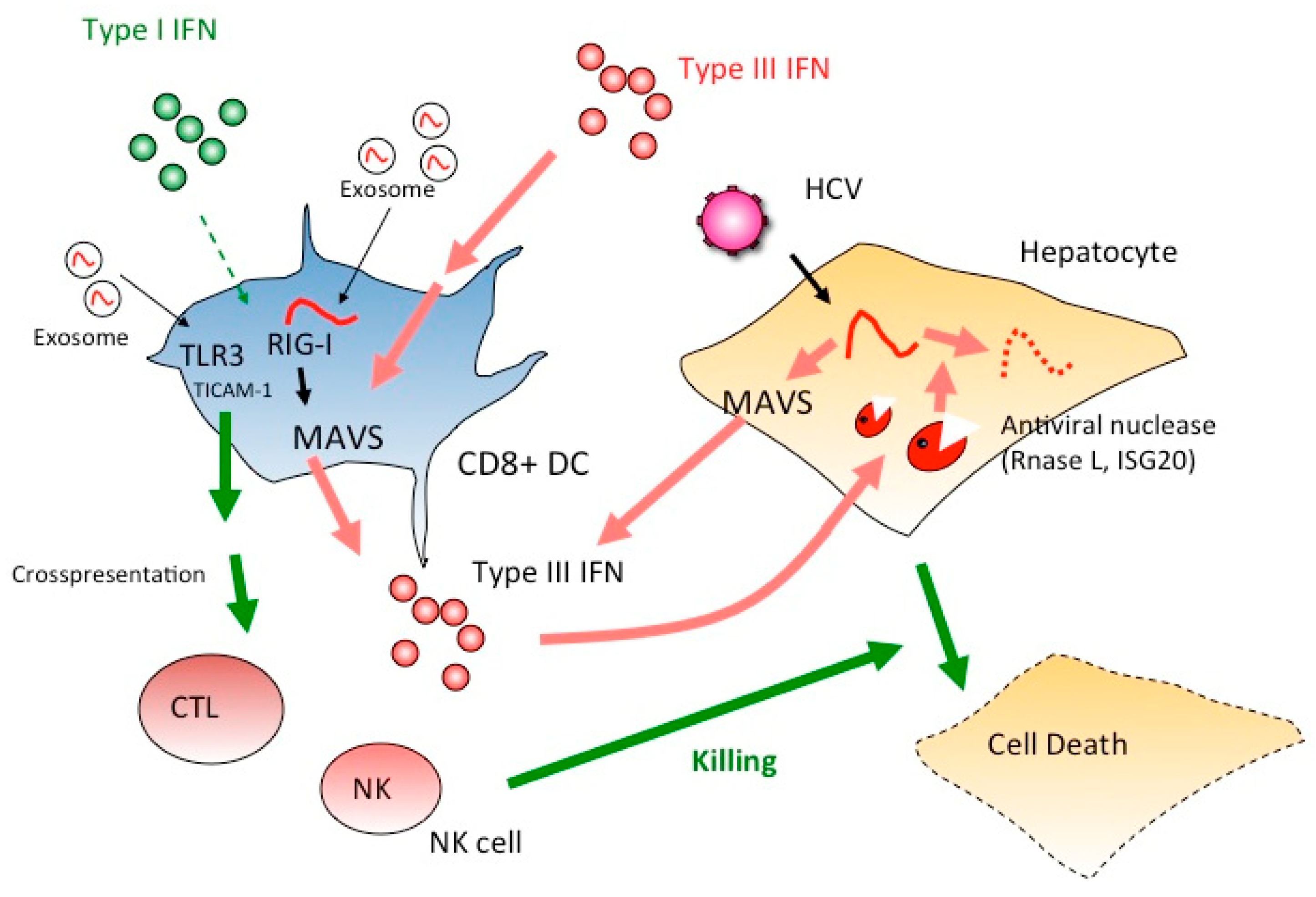
4. The IFN-Lambda Receptor in CD8+ DC Fails to Activate Cross-Priming or NK Activation

5. Extrinsically Added Hepatocyte Debris Containing HCV RNA Stimulates the TICAM-1 Pathway
6. TICAM-1 Signal Initiated by HCV RNA in Persistent Infection Induces Cellular Immunity
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Matsumoto, M.; Oshiumi, H.; Seya, T. Antiviral responses induced by the TLR3 pathway. Rev. Med. Virol. 2011, 21, 67–77. [Google Scholar]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [PubMed]
- Tatematsu, M.; Seya, T.; Matsumoto, M. Beyond dsRNA: Toll-like receptor 3 signalling in RNA-induced immune responses. Biochem. J. 2014, 458, 195–201. [Google Scholar] [PubMed]
- Lauterbach, H.; Bathke, B.; Gilles, S.; Traidl-Hoffmann, C.; Luber, C.A.; Fejer, G.; Freudenberg, M.A.; Davey, G.M.; Vremec, D.; Kallies, A.; et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J. Exp. Med. 2010, 207, 2703–2717. [Google Scholar] [PubMed]
- Oshiumi, H.; Funami, K.; Aly, H.H.; Matsumoto, M.; Seya, T. Multi-step regulation of interferon induction by hepatitis C virus. Arch. Immunol. Ther. Exp. (Warsz.) 2013, 61, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Funami, K.; Komori, A.; Yokoyama, T.; Aiba, Y.; Araki, A.; Takii, Y.; Ito, M.; Matsuyama, M.; Koyabu, M.; et al. Increased expression of Toll-like receptor 3 in intrahepatic biliary epithelial cells at sites of ductular reaction in diseased livers. Hepatol. Int. 2008, 2, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Oshiumi, H.; Azuma, M.; Kato, N.; Matsumoto, M.; Seya, T. IPS-1 is essential for type III IFN production by hepatocytes and dendritic cells in response to hepatitis C virus infection. J. Immunol. 2014, 192, 2770–2777. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Szabo, G. Dendritic cells in hepatitis C infection: Can they (help) win the battle? J. Gastroenterol. 2011, 46, 432–447. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, T.; Shingai, M.; Matsumoto, M.; Wakita, T.; Seya, T. Hepatitis C virus-infected hepatocytes extrinsically modulate dendritic cell maturation to activate T cells and natural killer cells. Hepatology 2008, 48, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Tsukiyama-Kohara, K.; Kohara, M. Tupaia belangeri as an experimental animal model for viral infection. Exp. Anim. 2014, 63, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Billerbeck, E.; de Jong, Y.; Dorner, M.; de la Fuente, C.; Ploss, A. Animal models for hepatitis C. Curr. Top. Microbiol. Immunol. 2013, 369, 49–86. [Google Scholar] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Tahara, H.; Ghivizzani, S.C. Viral vectors for gene therapy. Trends Biotechnol. 1998, 16, 35–40. [Google Scholar] [CrossRef]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1998, 6, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Owen, D.M.; Jiang, F.; Marcotrigiano, J.; Gale, M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 2008, 454, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Contag, C.H.; Ilves, H.; Johnston, B.H.; Kaspar, R.L. Small hairpin RNAs efficiently inhibit hepatitis C IRES—Mediated gene expression in human tissue culture cells and a mouse model. Mol. Ther. 2005, 12, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Shin, D.; Lee, H.; Ahn, B.Y.; Yoon, Y.; Kim, M. Targeted delivery of siRNA against hepatitis C virus by apolipoprotein A-Ibound cationic liposomes. J. Hepatol. 2009, 50, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Montoya, M.; Unger, H.; Alexopoulou, L.; Roy, P.; Haswell, L.E.; Al-Shamkhani, A.; Flavell, R.; Borrow, P.; e Sousa, C.R. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 2003, 424, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Nakai, M.; Seya, T.; Matsumoto, M.; Shimotohno, K.; Sakamoto, N.; Aly, H.H. The J6JFH1 Strain of Hepatitis C Virus Infects Human B-Cells with Low Replication Efficacy. Viral Immunol. 2014, 27, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stamataki, Z.; Shannon-Lowe, C.; Shaw, J.; Mutimer, D.; Rickinson, A.B.; Gordon, J.; Adams, D.H.; Balfe, P.; McKeating, J.A. Hepatitis C virus association with peripheral blood B lymphocytes potentiates viral infection of liver-derived hepatoma cells. Blood 2009, 113, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.L. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 4124–4130. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.; Zhang, J.; Flint, M.; Logvinoff, C.; Cheng-Mayer, C.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 2003, 100, 7271–7276. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Marukian, S.; Andrus, L.; Sheahan, T.P.; Jones, C.T.; Charles, E.D.; Ploss, A.; Rice, C.M.; Dustin, L.B. Hepatitis C virus induces interferon-λ and interferon-stimulated genes in primary liver cultures. Hepatology 2011, 54, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Gonzalez, V.D.; Li, Q.; Modi, A.A.; Chen, W.; Noureddin, M.; Rotman, Y.; Liang, T.J. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 2012, 142, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zhong, J.; Chisari, F.V. Inhibition of dsRNA-induced signaling in hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2006, 103, 8499–8504. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Ikeda, M.; Matsumoto, M.; Watanabe, A.; Takeuchi, O.; Akira, S.; Kato, N.; Shimotohno, K.; Seya, T. Hepatitis C virus core protein abrogates the DDX3 function that enhances IPS-1-mediated IFN-beta induction. PLoS ONE 2010, 5, e14258. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 2010, 8, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.E.; Schreckhise, H.; Khuu-Duong, K.; Henderson, K.; Rosler, R.; Storey, H.; Yao, L.; Liu, H.; Barahmand-Pour, F.; Sivakumar, P.; et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006, 44, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Marcello, T.; Grakoui, A.; Barba-Spaeth, G.; Machlin, E.S.; Kotenko, S.V.; MacDonald, M.R.; Rice, C.M. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006, 131, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.; Imanaka, N.; Marukian, S.; Dorner, M.; Liu, P.; Ploss, A.; Rice, C.M. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe 2014, 15, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Israelow, B.; Narbus, C.M.; Sourisseau, M.; Evans, M.J. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology 2014, 60, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Fiegen, A.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M. Insights into antiviral innate immunity revealed by studying hepatitis C virus. Cytokine 2015, 74, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.J.; Shiffman, M.L.; Zaman, A.; Yoffe, B.; de la Torre, A.; Flamm, S.; Gordon, S.C.; Marotta, P.; Vierling, J.M.; Lopez-Talavera, J.C.; et al. Phase 1b study of pegylated interferon lambda 1 with or without ribavirin in patients with chronic genotype 1 hepatitis C virus infection. Hepatology 2010, 52, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Marukian, S.; Jones, C.T.; Andrus, L.; Evans, M.J.; Ritola, K.D.; Charles, E.D.; Rice, C.M.; Dustin, L.B. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 2008, 48, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Laporte, J.; Bain, C.; Maurel, P.; Inchauspe, G.; Agut, H.; Cahour, A. Differential distribution and internal translation efficiency of hepatitis C virus quasispecies present in dendritic and liver cells. Blood 2003, 101, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Donada, C.; Crucitti, A.; Donadon, V.; Tommasi, L.; Zanette, G.; Crovatto, M.; Santini, G.F.; Chemello, L.; Alberti, A. Systemic manifestations and liver disease in patients with chronic hepatitis C and type II or III mixed cryoglobulinaemia. J. Viral Hepat. 1998, 5, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Sung, V.M.; Shimodaira, S.; Doughty, A.L.; Picchio, G.R.; Can, H.; Yen, T.S.; Lindsay, K.L.; Levine, A.M.; Lai, M.M. Establishment of B-cell lymphoma cell lines persistently infected with hepatitis C virus in vivo and in vitro: The apoptotic effects of virus infection. J. Virol. 2003, 77, 2134–2146. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Seya, T. Ubiquitin-mediated modulation of the cytoplasmic viral RNA sensor RIG-I. J. Biochem. 2012, 151, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Goutagny, N.; Fatmi, A.; de Ledinghen, V.; Penin, F.; Couzigou, P.; Inchauspe, G.; Bain, C. Evidence of viral replication in circulating dendritic cells during hepatitis C virus infection. J. Infect. Dis. 2003, 187, 1951–1958. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.N.Q.; MacParland, S.A.; Mulrooney, P.M.; Cooksley, H.; Naoumov, N.V.; Michalak, T.I. Hepatitis C virus persistence after spontaneous or treatment-induced resolution of hepatitis C. J. Virol. 2004, 78, 5867–5874. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Suzuki, T.; Miyashita, M.; Mifsud, E.J.; Leong, C.R.; Kohara, M.; Matsumoto, M.; Seya, T. Hepatitis B virus infection induces hepatic IFN-γ production in early infection, which causes cytoplasmic viral RNA instability via the DDX60 RNA exosome. Cell Rep. 2015. submitted. [Google Scholar]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Décembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Yoshio, S.; Kanto, T.; Kuroda, S.; Matsubara, T.; Higashitani, K.; Kakita, N.; Ishida, H.; Hiramatsu, N.; Nagano, H.; Sugiyama, M.; et al. Human blood dendritic cell antigen 3 (BDCA3)+ dendritic cells are a potent producer of interferon-λ in response to hepatitis C virus. Hepatology 2013, 57, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Takaki, H.; Oshiumi, H.; Matsumoto, M.; Seya, T. Dendritic cell subsets involved in type I IFN induction in mouse measles virus infection models. Int. J. Biochem. Cell Biol. 2014, 53C, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Takaki, H.; Takeda, M.; Tahara, M.; Shingai, M.; Oshiumi, H.; Matsumoto, M.; Seya, T. The MyD88 pathway in plasmacytoid and CD4+ dendritic cells primarily triggers type I IFN production against measles virus in a mouse infection model. J. Immunol. 2013, 191, 4740–4747. [Google Scholar] [CrossRef] [PubMed]
- Jomantaite, I.; Dikopoulos, N.; Kröger, A.; Leithäuser, F.; Hauser, H.; Schirmbeck, R.; Reimann, J. Hepatic dendritic cell subsets in the mouse. Eur. J. Immunol. 2004, 34, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, T.; Ebihara, T.; Okuno, M.; Okuda, Y.; Shingai, M.; Tsujimura, K.; Takahashi, T.; Ikawa, M.; Okabe, M.; Inoue, N.; et al. Antitumor NK activation induced by the Toll-like receptor 3-TICAM-1 (TRIF) pathway in myeloid dendritic cells. Proc. Natl. Acad. Sci. USA 2007, 104, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Seya, T.; Kasamatsu, J.; Azuma, M.; Shime, H.; Matsumoto, M. Natural killer cell activation secondary to innate pattern sensing. J. Innate Immun. 2011, 3, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Azuma, M.; Ebihara, T.; Oshiumi, H.; Matsumoto, M.; Seya, T. Cross-priming for antitumor CTL induced by soluble Ag + polyI:C depends on the TICAM-1 pathway in mouse CD11c(+)/CD8α(+) dendritic cells. Oncoimmunology 2012, 1, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Leong, C.R.; Oshiumi, H.; Deng, M.G.; Okamoto, M.; Takaki, H.; Matsumoto, M.; Seya, T. ISG20 inhibits HBV replication by degrading RNA of HBV origin. Sci. Rep. 2015. submitted. [Google Scholar]
- Leong, C.R.; Matsumoto, M.; Suzuki, T.; Oshiumi, H.; Seya, T. Nucleic acid sensors involved in the recognition of hepatitis B virus (HBV) in the liver-specific in vivo transfection mouse models-Pattern recognition receptors and sensors for HBV. Med. Sci. 2015, 3, 16–24. [Google Scholar]
- Zhu, H.; Dong, H.; Eksioglu, E.; Hemming, A.; Cao, M.; Crawford, J.M.; Nelson, D.R.; Liu, C. Hepatitis C Virus Triggers Apoptosis of a Newly Developed Hepatoma Cell Line through Antiviral Defense System. Gastroenterology 2007, 133, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Saitoh, S.; Suzuki, Y.; Kobayashi, M.; Tsubota, A.; Koida, I.; Arase, Y.; Fukuda, M.; Chayama, K.; Murashima, N.; et al. Disease progression and hepatocellular carcinogenesis in patients with chronic viral hepatitis: A prospective observation of 2215 patients. J. Hepatol. 1998, 28, 930–938. [Google Scholar] [CrossRef]
- Jacobs, J.L.; Coyne, C.B. Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol. 2013, 425, 5009–5019. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Gale, M., Jr. Regulation of hepatic innate immunity by hepatitis C virus. Nat. Med. 2013, 19, 879–888. [Google Scholar] [CrossRef] [PubMed]
- McCartney, S.; Vermi, W.; Gilfillan, S.; Cella, M.; Murphy, T.L.; Schreiber, R.D.; Murphy, K.M.; Colonna, M. Distinct and complementary functions of MDA5 and TLR3 in poly(I:C)-mediated activation of mouse NK cells. J. Exp. Med. 2009, 206, 2967–2976. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Lemon, S.M. Innate immune responses in hepatitis C virus infection. Semin. Immunopathol. 2013, 35, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Sawa, Y.; Arima, Y.; Ogura, H.; Kitabayashi, C.; Jiang, J.J.; Fukushima, T.; Kamimura, D.; Hirano, T.; Murakami, M. Hepatic interleukin-7 expression regulates T cell responses. Immunity 2009, 30, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakai, M.; Oshiumi, H.; Funami, K.; Okamoto, M.; Matsumoto, M.; Seya, T.; Sakamoto, N. Interferon (IFN) and Cellular Immune Response Evoked in RNA-Pattern Sensing During Infection with Hepatitis C Virus (HCV). Sensors 2015, 15, 27160-27173. https://doi.org/10.3390/s151027160
Nakai M, Oshiumi H, Funami K, Okamoto M, Matsumoto M, Seya T, Sakamoto N. Interferon (IFN) and Cellular Immune Response Evoked in RNA-Pattern Sensing During Infection with Hepatitis C Virus (HCV). Sensors. 2015; 15(10):27160-27173. https://doi.org/10.3390/s151027160
Chicago/Turabian StyleNakai, Masato, Hiroyuki Oshiumi, Kenji Funami, Masaaki Okamoto, Misako Matsumoto, Tsukasa Seya, and Naoya Sakamoto. 2015. "Interferon (IFN) and Cellular Immune Response Evoked in RNA-Pattern Sensing During Infection with Hepatitis C Virus (HCV)" Sensors 15, no. 10: 27160-27173. https://doi.org/10.3390/s151027160
APA StyleNakai, M., Oshiumi, H., Funami, K., Okamoto, M., Matsumoto, M., Seya, T., & Sakamoto, N. (2015). Interferon (IFN) and Cellular Immune Response Evoked in RNA-Pattern Sensing During Infection with Hepatitis C Virus (HCV). Sensors, 15(10), 27160-27173. https://doi.org/10.3390/s151027160